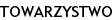Endokrynologia Pediatryczna, Tom 3 | Rok 2004 | Nr 1(6)
DOI: 10.18544/EP-01.03.01.0908
Adrenoleukodystrofia – patogeneza, diagnostyka i leczenie
Klinika Endokrynologii i Diabetologii Wieku Rozwojowego Akademii Medycznej im. Karola Marcinkowskiego w Poznaniu
Słowa kluczowe: adrenoleukodystrofia, adrenomieloneuropatia, niewydolność kory nadnerczy, przeszczep szpiku kostnego
Streszczenie
W opracowaniu zaprezentowane są patogeneza, objawy kliniczne, diagnostyka oraz możliwości leczenia adrenoleukodystrofii. Adrenoleukodystrofia sprzężona z chromosomem X jest dziedzicznym zaburzeniem peroksysomalnej β-oxydacji. Charakteryzuje się kumulacją kwasów tłuszczowych o bardzo długim łańcuchu (VLCFA), prowadzi do demielinizacji w ośrodkowym układzie nerwowym oraz zaburza steroidogenezę w korze nadnerczy i w jądrach. Ekspresja fenotypowa adrenoleukodystrofii jest bardzo zmienna, nawet w obrębie rodzeństwa z taką samą mutacją odpowiedzialnego genu. Najczęstszymi postaciami choroby są dziecięca mózgowa adrenoleukodystrofia oraz adrenomieloneuropatia. Diagnozę można potwierdzić, wykazując podwyższone stężenie VLCFA w osoczu albo dzięki analizie mutacji DNA. Możliwa jest diagnostyka prenatalna. U większości pacjentów występuje niewydolność kory nadnerczy, wymagająca sterydowej terapii substytucyjnej. Jeśli rozpoznanie choroby ma miejsce wcześnie, można rozważyć transplantację szpiku kostnego. Wiele innych metod terapeutycznych okazało się jak dotąd nieskutecznych albo nie zostały dostatecznie zbadane i zaakceptowane.
Wstęp
Adrenoleukodystrofia sprzężona z chromosomem X (X-ALD), znana dawniej jako leukodystrofia sudanofilna lub choroba Siemerlinga-Creutzfeldta, jest rzadką chorobą z pogranicza neurologii i endokrynologii. Zmiany patologiczne dotyczą przede wszystkim istoty białej OUN, kory nadnerczy oraz gonad męskich. U chorych występuje demielinizacja OUN, pierwotna niedoczynność nadnerczy oraz hipogonadyzm. Charakterystyczne, wydawałoby się, objawy mogą jednak pojawiać się w dowolnej kolejności i mieć różne nasilenie, co stanowi dla lekarza prawdziwe wyzwanie diagnostyczne. Badania przeprowadzone w Holandii wykazały, że czas upływający od wystąpienia pierwszych symptomów do rozpoznania X-ALD wynosił średnio 9,9 roku i wahał się od kilku miesięcy do 33 lat (!) [1]. Tymczasem długotrwały brak prawidłowej diagnozy nie pozostaje bez wpływu na stan pacjenta z adrenoleukodystrofią. Neurolog badający chorego z objawami klinicznymi przypominającymi np. stwardnienie rozsiane, nie powinien przeoczyć dyskretnych oznak niedoczynności kory nadnerczy, której dekompensacja może mieć skutki groźne dla życia. Endokrynolog, do którego trafia pacjent z pierwotną niewydolnością nadnerczy, powinien pamiętać, że u jej podłoża może leżeć nie tylko proces autoimmunologiczny czy wrodzony przerost nadnerczy, ale również jedna z rzadszych przyczyn, np. X-ALD. Co ciekawe, istnieją prace badawcze wykazujące, że adrenoleukodystrofia, której w podręcznikach endokrynologii nie poświęca się zwykle więcej niż parę zdań, może odpowiadać nawet za ponad 30% przypadków choroby Addisona [2]. Częstość występowania adrenoleukodystrofii ocenia się na co najmniej 1:100 000 [3], a różnice między poszczególnymi populacjami są nieznaczne.
Rys historyczny
Pierwsze opisy kliniczne odpowiadające adrenoleukodystrofii pochodzą z początków XX wieku: Schilder (1913, 1924), Simerling i Creutzfeldt (1923). Obecną nazwę choroby zaproponował Blaw w 1970 roku. W 1973 Schaumburg i Powers wykazali u pacjentów z adrenoleukodystrofią obecność charakterystycznych zmian ultrastrukturalnych w korze nadnerczy [4]. Trzy lata później Igarashi i wsp. zbadali skład chemiczny tych patologicznych struktur, co pozwoliło zaliczyć X-ALD do chorób związanych ze spichrzaniem lipidów [5]. W 1984 roku Singh i wsp. określili defekt metaboliczny leżący u podstawy zaburzeń i zlokalizowali go w peroksysomach [6]. Także na początku lat 80., dzięki opracowaniom Mosera i wsp. [7, 8] oraz Tsuji i wsp. [9], możliwa stała się specyficzna diagnostyka adrenoleukodystrofii z wykorzystaniem badań hodowanych fibroblastów skóry, krwinek czerwonych lub osocza. Stopniowo rozwijały się analizy genetyczne i molekularne. Dzięki licznym badaniom pojawiły się różnorodne propozycje terapeutyczne: leczenie dietą, terapia immunosupresyjna, przeszczepy szpiku. Pomimo tak znacznego postępu badawczego, adrenoleukodystrofia pozostaje chorobą w wielu punktach niejasną i niestety nieuleczalną.
Zaburzenia biochemiczne i podłoże genetyczne
Adrenoleukodystrofia jest genetycznie uwarunkowaną chorobą metaboliczną, sprzężoną z chromosomem X i dziedziczoną w sposób recesywny. Chorujący mężczyźni mogą przekazywać wadę tylko córkom, które stają się nosicielkami zmutowanego genu. U kobiet-heterozygot w każdej ciąży istnieje 50% prawdopodobieństwo przekazania mutacji potomstwu: córki będą wtedy również nosicielkami, u synów rozwinie się objawowa postać choroby.
Istotą schorzenia jest defekt peroksysomalnej β-oksydacji kwasów tłuszczowych o bardzo długim łańcuchu (VLCFA – very long chain fatty acids), powodujący ich kumulację w ustroju. Pod pojęciem „kwasy tłuszczowe o bardzo długim łańcuchu” rozumiemy tu nasycone, nierozgałęzione kwasy tłuszczowe o długości łańcucha wynoszącej przynajmniej 24 atomy węgla. Występują one normalnie w tkankach roślinnych i zwierzęcych, także w organizmach ssaków. W przebiegu adrenoleukodystrofii obserwuje się wielokrotny wzrost stężenia VLCFA w płynach ustrojowych i tkankach, szczególnie w istocie białej OUN, korze nadnerczy i w jądrach. Spichrzaniu ulega głównie kwas heksakozanowy, czyli cerolowy (C26:0), a także kwas tetrakozanowy, czyli lignocerynowy (C24:0).
Gen, którego mutacja odpowiada za rozwój X-ALD, znajduje się na długim ramieniu chromosomu X (Xq28) [10]. Posiada 10 eksonów i rozciąga się na długości 20 kb DNA. Jego prawidłowym produktem jest białko ALDP, należące do nadrodziny ABC (ATP binding casette) transporterów, zlokalizowane w błonie peroksysomów [11]. Pomimo licznych badań nie stwierdzono dotąd jednoznacznie, w jaki sposób białko ALDP umożliwia β-oksydację VLCFA. Być może bierze udział w transporcie VLCFA lub ich aktywnych pochodnych (VLCFA-koenzymA) do wnętrza peroksysomu. Najprawdopodobniej jednak białko ALDP ma znaczenie dla funkcjonowania syntetazy sprzęgającej VLCFA z koenzymem A.
Opisano już ponad 500 różnego typu mutacji genu ALD, wykrytych u chorych, m.in. delecje, przesunięcia ramki odczytu, mutacje nonsensowne i mutacje zmiany sensu [12]. Większość mutacji jest specyficzna dla danej rodziny. Najczęstsza z nich, delecja AG w pozycji 1801-1802 w eksonie 5 genu, występuje jedynie u ok. 12% dotkniętych rodzin [13].
Obraz kliniczny
W diagnostyce X-ALD zwraca uwagę wywiad rodzinny, obciążony w ok. 95% przypadków. Zdarza się jednak, że choroba, pojawiająca się w rodzinie od kilku pokoleń, nie była dotąd prawidłowo zdiagnozowana.
Dla obrazu klinicznego adrenoleukodystrofii typowe są nasilające się objawy neurologiczne, związane z demielinizacją istoty białej mózgu i rdzenia kręgowego. U pacjentów można zaobserwować postępujące upośledzenie funkcji intelektualnych. Chorzy chłopcy stają się roztargnieni, zaczynają mieć trudności w nauce szkolnej. Pogarsza się rozumienie mowy, pamięć, charakter pisma. Występują zaburzenia orientacji przestrzennej. Często zmienia się zachowanie, np. pojawia się agresja. Obserwuje się osłabienie słuchu, a później także wzroku pacjentów. Dołączają się zaburzenia równowagi i koordynacji ruchów, mogą występować drgawki. Objawy te prowadzą do otępienia, całkowitego kalectwa i śmierci chorego. Są również postacie choroby, w których zaburzenia neurologiczne nie postępują tak gwałtownie i ograniczają się raczej do spastycznego niedowładu kończyn dolnych i dysfunkcji zwieraczy [3].
Drugą główną cechą adrenoleukodystrofii jest pierwotna niedoczynność kory nadnerczy, która często wyraźnie wyprzedza rozwój objawów neurologicznych. Prawidłowa diagnoza, postawiona już na tym wczesnym etapie, umożliwia szybkie zastosowanie dostępnych metod terapeutycznych i daje szansę skuteczniejszej walki z chorobą. U pełnoobjawowych chorych niewydolność nadnerczy występuje w 70-90% przypadków. Jej ekspresja kliniczna waha się od dyskretnych przebarwień skóry, poprzez osłabienie z towarzyszącą uogólnioną hiperpigmentacją (ryc. 1a-c), aż po groźne dla życia przełomy korowo-nadnerczowe.
Badania morfologiczne i cytochemiczne przeprowadzone w latach 80. XX wieku przez Powersa i wsp. wykazały obecność dwułomnych, blaszkowatych wtrętów w cytoplazmie komórek wewnętrznej części strefy pasmowatej i siatkowatej kory nadnerczy [4]. Składnikiem patologicznych blaszek jest cholesterol zestryfikowany VLCFA [5], który nie może być prekursorem w syntezie hormonów steroidowych. W komórkach zawierających wtręty stwierdzono słabszą aktywność enzymów mitochondrialnych i mikrosomalnych. Dodatkowo, wysokie stężenia VLCFA mogą zaburzać strukturę i funkcję błon komórkowych. Whitcomb i wsp. [14], badając ten problem zauważyli, że dodanie do hodowli ludzkich komórek kory nadnerczy kwasu heksakozanowego lub tetrakozanowego w stężeniach odpowiadających ich poziomowi w osoczu chorych z X-ALD, nasila mikrolepkość błon komórkowych i osłabia wydzielanie kortyzolu w odpowiedzi na ACTH. Badacze doszli do wniosku, że nadmiar VLCFA w błonie komórki zaburza jej strukturę oraz upośledza dostępność receptorów dla ACTH.
W przebiegu adrenoleukodystrofii zaburzenie sterydogenezy dotyczy również jąder. W różnym stopniu nasilony pierwotny hipogonadyzm obecny jest u większości dorosłych pacjentów. U chorych może występować impotencja, zmiany owłosienia, ginekomastia. Czasami obserwuje się zaburzenia zstępowania jąder lub ich mniejszą objętość. Badania laboratoryjne osocza mogą wykazać niski podstawowy poziom testosteronu i DHEA-S, podwyższone stężenia LH i FSH oraz nieprawidłowe wyniki w testach stymulacyjnych z hCG i GnRH [15]. Zaburzenia te nie są jednak równoznaczne z bezpłodnością; zwłaszcza w łagodnych i późno rozwijających się formach choroby posiadanie potomstwa jest zwykle możliwe.
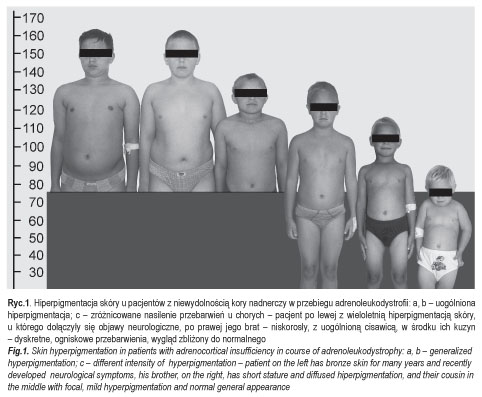
U chorych stwierdza się również charakterystyczny typ owłosienia głowy – włosy stają się przerzedzone i delikatne (ryc. 2.).
Postacie kliniczne
Zależnie od występujących objawów oraz momentu ich pojawienia się, można wyróżnić 6 postaci klinicznych choroby [16]: 1) mózgową postać dziecięcą; 2) mózgową postać młodzieńczą; 3) mózgową postać dorosłych; 4) adrenomieloneuropatię (AMN); 5) izolowaną niedoczynność kory nadnerczy; 6) postać asymptomatyczną.
Znakomitą większość przypadków stanowią mózgowa postać dziecięca i adrenomieloneuropatia. Zdarzają się również nietypowe neurologiczne warianty choroby, przypominające zanik oliwkowo–mostowo–móżdżkowy czy degenrację rdzeniowo-móżdżkową [3]. Odmiany mózgowe różnią się wiekiem wystąpienia pierwszych objawów: dziecięca – poniżej 10 roku życia, (ale właściwie nigdy przed 3 r.ż.), młodzieńcza – między 10 a 21 rokiem życia i dorosłych – powyżej 21 lat. Dla wszystkich trzech postaci charakterystyczna jest symetryczna, postępująca demielinizacja w obrębie półkul mózgowych. Zwykle rozpoczyna się ona w regionie ciemieniowo-potylicznym, a towarzyszy jej okołonaczyniowy naciek zapalny i uszkodzenie bariery krew–mózg. Objawy neurologiczne narastają bardzo szybko, prowadząc w ciągu 2-3 lat do zgonu chorego. Czasem obserwuje się przejście w przewlekły stan wegetatywny.
Odrębną postacią choroby jest AMN, która pojawia się u mężczyzn dorosłych – najczęściej młodych lub w wieku średnim. Jej objawy mogą postępować powoli, nawet przez dziesięciolecia. Dotknięty jest przede wszystkim rdzeń kręgowy, a zmiany demielinizacyjne rzadko łączą się z procesem zapalnym. Występuje również neuropatia obwodowa. Pacjenci skarżą się głównie na postępujące osłabienie i niedowład kończyn dolnych oraz zaburzenia czynności zwieraczy i impotencję. Podobne objawy kliniczne oraz zbliżony obraz rezonansu magnetycznego głowy powodują, że ta odmiana X-ALD mylona jest nierzadko z najczęstszą chorobą demielinizacyjną OUN, czyli ze stwardnieniem rozsianym. W wykluczeniu fałszywego rozpoznania SM pomagają badania immunologiczne płynu mózgowo-rdzeniowego oraz ocena wzrokowych potencjałów wywołanych (VEP). W adrenomieloneuropatii po latach może dojść do gwałtownego nasilenia zaburzeń neurologicznych, przypominającego postać mózgową.
Wszystkim powyższym postaciom adrenoleukodystrofii towarzyszy często niedoczynność kory nadnerczy. Zdarza się również, iż pierwotna niewydolność nadnerczy jest jedynym uchwytnym przejawem klinicznym X-ALD, a stan neurologiczny chorego nie budzi zastrzeżeń. Pacjent trafia wtedy pod opiekę endokrynologa, który powinien mieć świadomość, że izolowana niedoczynność kory nadnerczy może być jedną z postaci X-ALD. Dlatego przy najmniejszym podejrzeniu zaburzeń neurologicznych lub przy niejasnym wywiadzie rodzinnym, obciążonym bliżej nieokreślonymi schorzeniami układu nerwowego, celowe jest poszerzenie diagnostyki o tę rzadką przyczynę dysfunkcji kory nadnerczy.
Czasem, podczas badań rodziny chorego, wykryte zostają całkiem bezobjawowe przypadki adrenoleukodystrofii. Dotyczą osób, u których występuje mutacja genu oraz podwyższenie osoczowego poziomu VLCFA, ale pod względem neurologicznym oraz endokrynologicznym ich stan jest prawidłowy. Niestety, długoterminowa obserwacja wykazuje, że zarówno postać asymptomatyczna X-ALD, jak i izolowana niedoczynność kory nadnerczy u pacjentów z X-ALD rozwijają się z czasem w pełnoobjawową postać choroby, zwykle o charakterze adrenomieloneuropatii [16]. Istnieją jednak udokumentowane przypadki nawet ponadsiedemdziesięcioletnich chorych, u których objawy adrenoleukodystrofii ograniczyły się do niewydolności nadnerczy [3].
Opisywano także pacjentów, u których jedynymi objawami X-ALD były cechy hipogonadyzmu i dlatego niektórzy proponują wyróżnienie jeszcze jednej postaci choroby – izolowanej niedoczynności gonad [15].
Przy omawianiu obrazów klinicznych adrenoleukodystrofii warto również wspomnieć o możliwości wystąpienia zmian typu łagodnej lub umiarkowanie nasilonej adrenomieloneuropatii u kobiet, nosicielek zmutowanego genu. Przede wszystkim są to objawy spastycznej paraparezy, pojawiające się w starszym wieku u ok. 20% heterozygot. Niedoczynność kory nadnerczy, podobnie jak postacie z zajęciem mózgu, stwierdza się u nich bardzo rzadko [17].
Dużym problemem w X-ALD jest znaczna nieprzewidywalność przebiegu choroby. Nasilenie objawów nie wiąże się z poziomem VLCFA w organizmie. Brak również korelacji między genotypem a fenotypem, tzn. dany typ mutacji może powodować różne objawy kliniczne [18, 19]. Z tego powodu w jednej rodzinie występują często odmienne postacie X-ALD, nawet wśród rodzeństwa. Istnieją różne hipotezy, dotyczące przyczyn tego faktu. Jedna z nich nawiązuje do budowy molekularnej białek z nadrodziny ABC transporterów. Łączą się one w dimery, tworzące kanały przezbłonowe. Każdy monomer posiada domenę hydrofobową, która przenika błonę i prawdopodobnie odpowiada za specyficzność transportową powstałego kanału, oraz domenę cytoplazmatyczną o bardzo konserwatywnej sekwencji, z którą wiąże się ATP. Takie dimeryczne kanały mogą składać się z dwóch identycznych białek (homodimer) lub dwóch różnych cząsteczek z nadrodziny ABC transporterów (heterodimer) [16]. W błonie peroksysomów, oprócz białka ALDP, wykryto także trzy inne ABC transportery: białka ALDR, PMP70 i PMP70R, charakteryzujące się wysoką homologią z białkiem ALDP [20]. Przypuszcza się, że różnice w interakcji ALDP i jego homologów mogą wpływać na ekspresję fenotypową choroby. Badania in vitro wykazały, że białko ALDR może zastępować ALDP, przywracając zdolność degradacji VLCFA fibroblastom chorych z adrenoleukodystrofią [21].
Są również teorie zakładające, że na wystąpienie danej postaci X-ALD może wpływać jakiś dodatkowy gen modyfikujący [22]. Ponieważ na poziomie tkankowym główną cechą różnicującą gwałtownie przebiegające formy mózgowe od łagodniejszej adrenomieloneuropatii jest obecność lub brak odczynu zapalnego w mózgu, niektórzy badacze sugerują, że ewentualny gen modyfikujący mógłby działać przez modulowanie odpowiedzi zapalnej. Dotychczasowe prace, koncentrujące się na ekspresji TNFα oraz innych cytokin nie potwierdziły jednak tej hipotezy [23].
Mówi się także o wpływie bliżej nieokreślonych czynników środowiskowych na przebieg adrenoleukodystrofii, czego dowodem mają być przypadki bliźniąt jednojajowych, u których wystąpiły odmienne postacie choroby [24, 25].
Diagnostyka
Rozstrzygające dla rozpoznania adrenoleukodystrofii jest stwierdzenie podwyższonego stężenia VLCFA w osoczu [8]. Zwykle bada się poziom kwasu heksakozanowego (C26:0) oraz stosunek tego kwasu do kwasu dokozanowego, czyli behenowego (C26:0/C22:0), a także proporcję między kwasem tetrakozanowym a dokozanowym (C24:0/C22:0). Oznaczenia takie wykonywane są w niewielu specjalistycznych laboratoriach, między innymi w Pracowni Lipidów Zakładu Diagnostyki Laboratoryjnej w Centrum Zdrowia Dziecka w Warszawie. Każde laboratorium opracowuje własny zakres prawidłowych stężeń VLCFA, a wynik badania nadesłanych próbek opatruje komentarzem wyjaśniającym, czy mieści się on w granicach normy, czy jest patologiczny. Pracownie starają się również tak doprecyzować zakresy norm, aby na podstawie badania poziomu VLCFA móc wyróżnić nie tylko osoby chore i zdrowe, ale także kobiety-nosicielki zmutowanego genu. Metoda ta pozwala zidentyfikować właściwie wszystkich chorych, a u heterozygot-nosicielek ma 85% czułość diagnostyczną. Podwyższoną zawartość VLCFA wykazuje się również w hodowanych fibroblastach skóry [7] i w czerwonych krwinkach [9], choć nie są to metody rutynowe.
Diagnostykę mogą wspomagać badania molekularne. Wykonuje je tylko kilka wyspecjalizowanych placówek na świecie, są one bardzo kosztowne, a przede wszystkim wymagają uprzedniej identyfikacji mutacji występującej w danej rodzinie. Utrudnieniem jest tutaj wspomniana wcześniej znaczna różnorodność spotykanych mutacji. Dlatego badania te znajdują zastosowanie głównie przy określaniu nosicielstwa u krewnych płci żeńskiej, u których poziom VLCFA mieści się w normie [26].
Zarówno badanie poziomu VLCFA, jak i analizy molekularne mogą być wykorzystywane w diagnostyce prenatalnej X-ALD [27]. Materiałem do badań są hodowle komórek uzyskanych z płynu owodniowego (15-17 tydzień ciąży) lub z biopsji kosmówki (9-11 tydzień ciąży). Analiza DNA komórek płodowych wydaje się metodą pewniejszą niż ocena stężenia VLCFA, na temat której istnieją także doniesienia o wynikach fałszywie negatywnych [28].
Ważnym etapem badań chorego z adrenoleukodystrofią jest ocena czynności kory nadnerczy. U pacjentów z pierwotną niewydolnością kory nadnerczy wykrywa się podwyższony poziom ACTH w osoczu oraz obniżone stężenie kortyzolu w surowicy. Kora nadnerczy nie reaguje na stymulację Synacthenem. Czasami upośledzenie hormonalnej czynności kory nadnerczy charakteryzuje się wartościami kortyzolemii mieszczącymi się w dolnych granicach normy. Objawy kliniczne mogą być wtedy nikłe, ale w teście z Synacthenem brak jest prawidłowej odpowiedzi. W takich przypadkach niedoczynność kory nadnerczy może objawić się nagle w sytuacji stresowej, podczas operacji czy infekcji, w postaci ostrego kryzysu nadnerczowego [29].
Od strony neurologicznej duże znaczenie dla pacjentów z X-ALD ma ocena zmian w mózgu na podstawie rezonansu magnetycznego głowy. U chorych z postacią mózgową stwierdza się postępujący, symetryczny proces demielinizacyjny istoty białej, głównie okolic ciemieniowo-potylicznych. Natomiast w około 50% przypadków adrenomieloneuropatii rezonans mózgu nie wykazuje patologii [30].
Leczenie
Jak dotąd nie dysponujemy skuteczną metodą leczenia adrenoleukodystrofii. Jeszcze 10-20 lat temu duże nadzieje wiązano z terapią dietą. Przeprowadzone badania wykazały, że nadmiar VLCFA w mózgu chorych jest przynajmniej częściowo pochodzenia egzogennego [31]. Jednakże wyraźne ograniczenie spożycia pokarmów bogatych w kwas heksakozanowy nie przyniosło oczekiwanego spadku jego stężenia w organizmie chorych, ponieważ źródłem nadmiaru VLCFA w ustroju jest nie tylko dieta, ale w jeszcze większym stopniu synteza endogenna. Kwasy tłuszczowe o długości łańcucha przekraczającej 16 atomów węgla powstają w układach elongacyjnych w obrębie frakcji mikrosomalnej, a także w mitochondriach. Doświadczalnie wykazano, iż dodanie jednonienasyconego kwasu olejowego (C18:1;9) obniża poziomy VLCFA w hodowli fibroblastów skóry pacjentów z X-ALD [32]. Stwierdzono także, że za wydłużanie nasyconych kwasów tłuszczowych, np. behenowego (C22:0), jak również ich jednonienasyconych odpowiedników, np. kwasu erukowego (C22:1), odpowiada ten sam enzym [33]. Stąd pomysł, aby dietę eliminacyjną połączyć z podawaniem jednonienasyconych kwasów tłuszczowych w celu kompetycyjnego blokowania dostępu do mikrosomalnego układu elongacyjnego dla VLCFA. Ostatecznie zdecydowano się na stosowanie trójoleinianu (kw. olejowy C18:1;9) glicerolu (GTO) i trójerukanu (kw. erukowy C22:1;13) glicerolu (GTE) w proporcji 4:1 [34]. Mieszaninę tę nazywa się często „olejem Lorenza”, ponieważ do jej opracowania przyczynili się państwo Odone, rodzice chorego chłopca o tym imieniu. U większości pacjentów podawanie oleju Lorenza powodowało w ciągu kilku tygodni obniżenie stężenia kwasu heksakozanowego w osoczu do normalnego poziomu przy stosunkowo niewielkich efektach ubocznych w postaci umiarkowanej trombocytopenii [35]. Ogólnie zachęcające wyniki stosowania tej metody sprawiły, że rozpoczęto leczenie kilkuset pacjentów na całym świecie. Względy etyczne uniemożliwiły przeprowadzenie obiektywnych, randomizowanych badań w celu oceny skuteczności terapii. Niestety, obecnie przyjmuje się, że u chorych z występującymi już zaburzeniami neurologicznymi wpływ oleju Lorenza na postęp objawów jest nieznaczny lub w ogóle nie występuje, ponieważ kwas erukowy, główny składnik odpowiedzialny za obniżanie poziomu VLCFA, nie przenika przez barierę krew–mózg [36]. Dlatego olej Lorenza, który wyraźnie obniża stężenie VLCFA w osoczu, nie wpływa już tak korzystnie na ich poziom w mózgu i niestety nie hamuje postępu choroby. Co więcej, w świetle nowszych prac nadzieje wiązane z zastosowaniem mieszaniny GTO:GTE u chorych bez objawów z ośrodkowego układu nerwowego okazują się również złudne [37].
Obecnie jedyną powszechnie akceptowaną i zalecaną metodą terapeutyczną w adrenoleukodystrofii są allogeniczne przeszczepy szpiku kostnego. Pierwsze próby, przeprowadzone u pacjentów w zaawansowanym stadium choroby, zakończyły się niepowodzeniem: zaburzenia neurologiczne postępowały, nawet pomimo uzyskiwanego spadku stężenia VLCFA w osoczu [38]. Dopiero na początku lat 90. XX wieku pojawiły się bardziej optymistyczne doniesienia o skuteczności tej metody w przypadku zastosowania jej u chorych z niewielkimi zaburzeniami neurologicznymi i nieznacznymi zmianami w obrazie MRI mózgu. Po około pół roku od zabiegu stan pacjentów ulegał stabilizacji, a nawet zaczynał się poprawiać [39]. Istnieją już kilkuletnie obserwacje potwierdzające dobre efekty przeszczepów szpiku w przebiegu adrenoleukodystrofii. Najprawdopodobniej do poprawy dochodzi dzięki skutecznemu obniżeniu poziomu VLCFA w mózgu. Z przeszczepionego zdrowego szpiku pochodzą komórki zdolne do metabolizowania VLCFA, między innymi komórki zasiedlające OUN jako mikroglej. Prawidłowe komórki prawdopodobnie degradują nadmiar VLCFA w mózgu i hamują proces demielinizacji [16]. Z tą hipotezą koresponduje fakt, że poprawa stanu klinicznego rozpoczyna się po około 6 miesiącach od przeszczepu, co odpowiadałoby powolnej wymianie mikrogleju. Teorii tej nie można jednoznacznie potwierdzić, ponieważ nie ma sposobu nieinwazyjnego przyżyciowego pomiaru stężenia VLCFA w mózgu, a jak wiadomo z badań sekcyjnych [36], ich poziomy w osoczu i w obrębie tkanki ośrodkowego układu nerwowego nie koniecznie są skorelowane. Po przeszczepie szpiku osoczowy poziom VLCFA z reguły, choć nie zawsze, obniża się. Nie ma jednak bezpośredniego związku między zmianami stężeń VLCFA a stopniem poprawy klinicznej [40].
W chwili obecnej przeszczepy stosuje się u chorych z nieznacznymi objawami neurologicznymi i niewielkimi zmianami w MRI mózgu, o ile znajdzie się dla nich odpowiedni dawca. Nie zaleca się zabiegów u pacjentów z zaawansowanymi i szybko postępującymi zmianami w układzie nerwowym, ponieważ uzyskiwane wyniki nie są pozytywne, a stan chorych często ulega wręcz pogorszeniu [40]. Zwykle unika się także stosowania tej obciążającej procedury w przypadkach bezobjawowych z prawidłowym obrazem MRI mózgu i bez obciążonego wywiadu rodzinnego. Przeszczepy szpiku nadal wiążą się z wyraźnym ryzykiem dla życia pacjenta, natomiast chorzy tacy mają ponad 50% szans, że nie dojdzie u nich do rozwoju ciężkiej postaci mózgowej X-ALD [16]. Zaleca się wtedy systematyczną obserwację pacjenta, co 6-12 miesięcy, aby szybko wykrywać pierwsze oznaki postępu choroby i kwalifikować chorych do transplantacji. Właśnie dlatego dużą rolę odgrywa wczesne rozpoznanie adrenoleukodystrofii – już na etapie izolowanej niewydolności kory nadnerczy. Warto dodać, że przeszczepy szpiku u chłopców z adrenoleukodystrofią wykonywane są również w Polsce i jak na razie jest to jedyna metoda terapii X-ALD dostępna w naszym kraju.
Wśród proponowanych metod terapeutycznych wspomina się również o leczeniu immunosupresyjnym, którego celem miałoby być osłabienie odpowiedzi zapalnej w mózgu chorych z gwałtownie przebiegającymi postaciami X-ALD. Jednakże przeprowadzone dotąd badania z cyklofosfamidem i prednizonem, okazały się nieskuteczne w zapobieganiu postępowi objawów neurologicznych [41].
Pewne podobieństwo między adrenoleukodystrofią a stwardnieniem rozsianym dało podstawę dla prób zastosowania interferonu β w celu zahamowania rozwoju zaburzeń neurologicznych. Niestety, istnieją już doniesienia o braku pozytywnych efektów tej metody [42].
Próbowano również podawać chorym klofibrat [43], ale poza obniżeniem stężenia VLCFA w osoczu, nie powodował on korzystnych zmian klinicznych. Obecnie prowadzone są badania z zastosowaniem statyn lub 4-fenylobutyratu. Pojawiły się także doniesienia o korzystnym wpływie metabolitów testosteronu: 5α-androstan-17β-ol-3-onu (5α-dihydrotestosteron, 5α-DHT) i 5α-androstan-3α,17β-diolu na poziom VLCFA w hodowanych fibroblastach skóry [44], jednak są to zaledwie efekty badań laboratoryjnych i nie wiadomo, jakie będzie ich przyszłe odzwierciedlenie kliniczne.
Być może w przyszłości rozwój terapii genowej stanie się szansą opanowania choroby. Adrenoleukodystrofia wydaje się dobrym kandydatem dla takiej drogi leczenia: znany jest gen, którego mutacja leży u podstaw schorzenia, a diagnoza możliwa jest teoretycznie na długo przed rozwojem ciężkich objawów neurologicznych. Wprowadzenie skutecznego leczenia w momencie, gdy choroba nie daje jeszcze objawów, mogłoby zapobiegać ich rozwojowi i ratować życie pacjenta. Istnieją już badania stanowiące pierwszy krok w tym kierunku – za pomocą retrowirusów dokonano transfekcji hodowlanych fibroblastów chorego z X-ALD prawidłowym cDNA genu kodującego białko ALDP, uzyskując w efekcie poprawę upośledzonego metabolizowania VLCFA [45]. Jest to jednocześnie dowód, że to właśnie defekt białka ALDP jest przyczyną adrenoleukodystrofii.
W walkę z adrenoleukodystrofią angażują się instytucje publiczne i prywatne na całym świecie. Powstają różne organizacje, których zadaniem jest zbieranie danych o chorobie, propagowanie problematyki adrenoleukodystrofii w środowiskach naukowych, wspieranie inicjatyw badawczych oraz zbiórka funduszy na te cele, np. The STOP ALD Foundation, nad której działaniem czuwa rada naukowa złożona z najwybitniejszych specjalistów zajmujących się adrenoleukodystrofią. Na stronach internetowych The STOP ALD Foundation (www.stopald.org) można znaleźć fakty dotyczące samej choroby, jak również informacje o istniejących metodach terapeutycznych oraz obiecujących kierunkach badań z wykorzystaniem najnowszych technologii biologicznych. Wśród obecnie dostępnych sposobów leczenia zwraca uwagę przeszczep krwi pępowinowej (umbilical cord blood transplant – UCBT). Podobnie jak w przypadku transplantacji szpiku, celem zabiegu jest dostarczenie pacjentowi zdrowych komórek macierzystych (stem-cells); dodatkowe zalety tej metody to mniejsze wymagania dotyczące dopasowania biorcy i dawcy, a przez to krótszy czas oczekiwania na przeszczep, oraz mniejsze ryzyko reakcji przeszczepu przeciw gospodarzowi (GvHD). Z kolei słabą stroną UCBT jest często dłuższy proces przyjmowania się nowych komórek, a co za tym idzie, dłuższy okres immunosupresji i odpornościowej bezbronności chorego. Zarówno BMT, jak i UCBT są przeszczepami allogenicznymi, tymczasem największe nadzieje wiąże się ostatnio z zabiegami autologicznymi, wykorzystującymi możliwości terapii genowej. Idea takich procedur polega na pobraniu własnych komórek pacjenta, włączeniu do ich DNA prawidłowej sekwencji kodującej białko ALDP, namnożeniu tych komórek i ponownym wprowadzeniu ich do organizmu chorego. Zastosowanie tego typu metody pozwoliłoby wyeliminować znaczną część ryzyka związanego z przeszczepami allogenicznymi. Dodatkowo możliwe byłoby leczenie również tych chorych, dla których nie udało się znaleźć odpowiedniego dawcy. Według The STOP ALD Foundation zachęcające wyniki doświadczeń przeprowadzonych na transgenicznych myszach świadczą, iż warto pracować nad udoskonaleniem i standaryzacją tej metody. Trwają już przygotowania, które umożliwić mają podjęcie prób klinicznych.
Niestety, aktualnie wobec braku leczenia przyczynowego X-ALD, opieka nad chorymi ogranicza się do postępowania objawowego i podtrzymującego. Wskazane są regularne kontrole neurologiczne i endokrynologiczne. U pacjentów z niewydolnością kory nadnerczy należy wprowadzić substytucję hormonalną. Zazwyczaj wystarcza podawanie odpowiednich dawek hydrokortyzonu. O prawidłowym doborze dawek świadczy poprawa stanu klinicznego oraz wyniki okresowych kontrolnych badań laboratoryjnych: dobowe profile kortyzolemii i wydalanie kortyzolu z moczem. Czasem konieczna jest również substytucja mineralokortykosteroidów, choć warstwa kłębkowata wydaje się najmniej dotknięta przez proces patologiczny w korze nadnerczy. Nie jest znane wyjaśnienie takiej wybiórczości zmian. Leczenie niedoczynności kory nadnerczy nie wpływa na stan neurologiczny pacjenta, jednak przypadkowe przeoczenie jej objawów i brak odpowiedniej terapii mogą być bezpośrednią przyczyną zgonu chorego z adrenoleukodystrofią, np. w przebiegu innych dodatkowych chorób.
Leczenie objawowe obejmuje także środki przeciwdrgawkowe i obniżające napięcie mięśniowe. Wraz z pogarszaniem się stanu pacjenta konieczna staje się właściwa pielęgnacja. Ze względu na dramatyczny przebieg choroby, pacjenci oraz ich bliscy powinni być otoczeni opieką psychologiczną. W niektórych krajach istnieją specjalne grupy wsparcia, tworzone przez samych chorych i ich rodziny przy udziale lekarzy i psychologów, np. ALD Family Support Trust w Wielkiej Brytanii, Association Européenne contre les Leucodystrophies we Francji czy United Leukodystrophy Foundation w USA (www.ulf.org). Wskazane jest również poradnictwo genetyczne dla rodzin, w celu wyjaśnienia sposobu dziedziczenia adrenoleukodystrofii i zmniejszenia związanego z tym ryzyka.
Adrenoleukodystrofia nie należy do chorób rozpowszechnionych, warto jednak znać jej cechy charakterystyczne i pamiętać o niej w diagnostyce różnicowej schorzeń neurologicznych i endokrynologicznych. Endokrynolog, stykający się z przypadkami pierwotnej niedoczynności kory nadnerczy, nie powinien ograniczać rozpoznania do prostego stwierdzenia choroby Addisona. Obecnie, wobec spadku znaczenia czynników zakaźnych w etiologii tego schorzenia, relatywnie wzrosła częstość występowania przyczyn pozostałych, choć oczywiście najważniejszy pozostaje proces autoimmunologiczny. Dobrze, aby w praktycznym algorytmie postępowania diagnostycznego znalazło się badanie autoprzeciwciał skierowanych przeciwko enzymom steroidogenezy nadnerczowej, np. przy zastosowaniu precyzyjnych metod oznaczania przeciwciał przeciw 21-hydroksylazie sterydowej [46]. U chorych płci męskiej, u których niedoczynność kory nadnerczy występuje rodzinnie wraz z zaburzeniami neurologicznymi, albo u pacjentów z wywiadem rodzinnym obciążonym chorobami neurologicznymi, w tym oczywiście X-ALD, należy poszerzyć diagnostykę o ocenę stężenia VLCFA w osoczu. W przypadku patologicznego wyniku tych oznaczeń powinno się dążyć do zdiagnozowania pozostałych członków rodziny pod kątem choroby lub nosicielstwa mutacji. U pacjentów zdiagnozowanych na wczesnym etapie zaburzeń do rozważenia pozostaje możliwość wykonania transplantacji szpiku kostnego.
Piśmiennictwo
1. Van Geel B.M., Assies J., Haverkort E.B. et al.; Delay in diagnosis of X-linked aderenoleukodystrophy; Clin. Neurol. Neurosurg. 1993:95, 115-120
2. Laureti S., Casucci G., Santeusanio F. et al.; X-linked aderenoleukodystrophy is a frequent cause of idiopathic Addison’s disease in young adult male patients; J. Clin. Endocr. Metab. 1996:81, 470-474
3. Van Geel B.M., Assies J., Wanders R.J.A., Barth P.G.; X-linked aderenoleukodystrophy: clinical presentation, diagnosis and therapy; J. Neurol. Neurosurg. Psychiatry 1997:63, 4-14
4. Powers J.M., Schaumburg H.H.; The adrenal cortex in adreno-leukodystrophy; Arch. Pathol. 1973: 96, 305-310
5. Igarashi M., Schaumburg HH., Powers J. et al.; Fatty acid abnormality in aderenoleukodystrophy; J. Neurochem. 1976:26, 851-860
6. Singh I., Moser A.E., Moser H.W., Kishmoto Y.; Aderenoleukodystrophy: impaired oxidation of very long chain fatty acids in white blood cells, cultured skin fibroblasts and amniocytes; Pediatr. Res. 1984:18, 286-290
7. Moser H.W., Moser A.B., Kawamura N. et al.; Aderenoleukodystrophy: elevated C-26 fatty acid in cultured skin fibroblasts; Ann. Neurol. 1980:7, 542-549
8. Moser H.W., Moser A.B., Frayer K.K. et al.; Aderenoleukodystrophy: increased plasma content of saturated very long chain fatty acids; Neurology 1981:31, 1241-1246
9. Tsuji S., Suzuki M., Ariga T. et al.; Abnormality of long-chain fatty acids in erythrocyte membrane sphingomyelin from patients with adrenoleukodystrophy; J. Neurochem. 1981:36, 1046-1049
10. Migeon B.R., Moser H.W., Moser A.B. et al.; Aderenoleukodystrophy: evidence for X-linkage, inactivation and selection favoring the mutant allele in heterozygous cells; Proc. Natl. Acad. Sci. USA 1981:78, 5066-5070
11. Mosser J., Lutz Y., Stoeckel M.E. et al.; The gene responsible for aderenoleukodystrophy encodes for a peroxisomal membrane protein; Hum. Mol. Genet. 1994:3, 265-271
12. ; www.x-ald.nl;
13. Kemp S., Ligtenberg M.J., van Geel B.M. et al.; Identification of two base pair deletion in five unrelated families with aderenoleukodystrophy: a possible hot spot for mutations; Biochem. Biophys. Res. Commun. 1994:202, 647-653
14. Whitcomb R.W., Linehan W.M., Knazek R.A.; Effects of long-chain, saturated fatty acids on membrane microviscosity and adrenocorticotropin responsiveness of human adrenocortical cells in vitro; J. Clin. Invest. 1988:81, 185-188
15. Assies J., van Geel B.M., Gooren L.J.G., Barth P.G.; Signs of testicular insufficiency in adrenomyeloneuropathy and neurologically asymptomatic X-linked adrenoleukodystrophy: a retrospective study; Int. J. Andrology 1997:20, 315-321
16. Moser H.W.; Aderenoleukodystrophy: phenotype, genetics, pathogenesis and therapy; Brain 1997:120, 1485-1508
17. Moser H.W., Moser A.B., Naidu S. et al.; Clinical aspects of adrenoleukodystrophy and adrenomyeloneuropathy; Dev. Neurosci. 1991:13, 254-261
18. Kok F., Neumann S., Sarde C. et al.; Mutational analysis of patients with X-linked adrenoleukodystrophy; Hum. Mutat. 1995:6, 104-115
19. Krasemann E.W., Meier V., Korenke G.C. et al.; Identification of mutations in the ALD-gene of 20 families with adrenoleukodystrophy/adrenomyeloneuropathy; Hum. Genet. 1996:97, 194-197
20. Shani N., Steel G., Dean M. et al.; Four half ABC transporters may heterodimerize in the peroxisome membrane (abstract); Am. J. Hum. Genet. 1996:59, A42
21. Netik A., Forss-Petter S., Holzinger A. et al.; ALD-related protein can compensate functionally for ALD protein deficiency (X-ALD): implications for therapy; Hum. Molec. Genet. 1999:8, 907-913
22. Maestri N.E., Beaty T.H.; Predictions of a 2-locus model for disease heterogeneity: applications to adrenoleukodystrophy; Am. J. Med .Genet. 1992:44, 576-582
23. McGuinness M.C., Griffin D.E., Raymond G.V. et al.; Tumor necrosis factor-α and X-linked adrenoleukodystrophy; J. Neuroimmunol. 1995:61, 161-169
24. Korenke G.C., Fuchs S., Krasemann E. et al.; Cerebral adrenoleukodystrophy (ALD) in only one of monozygotic twins with an identical ALD genotype; Ann. Neurol. 1996:40, 254-257
25. Di Rocco M., Doria-Lamba L., Caruso U.; Monozygotic twins with X-linked adrenoleukodystrophy and different phenotypes; Ann. Neurol. 2001:50, 424
26. Notarangelo L.D., Parolini O., Baiguini G. et al. ; Carrier detection in X-linked adrenoleukodystrophy by determination of very long chain fatty acids and by linkage analysis; Eur. J. Pediatr. 1992:151, 761-763
27. Boué J., Oberle I., Heilig R. et al.; First trimester prenatal diagnosis of adrenoleukodystrophy by determination of very long chain fatty acids and by linkage analysis to a DNA probe; Hum. Genet. 1985:69, 272-274
28. Gray R.G.F., Green A., Cole T. et al.; A misdiagnosis of X-linked adrenoleukodystrophy in cultured chorionic villus cells by the measurement of very long chain fatty acids; Prenat. Diagn. 1995:15, 486-490
29. Luciani G.B., Pessotto R., Mazzucco A.; Adrenoleukodystrophy presenting as postperfusion syndrome; New. Eng. J. Med. 1997:336, 731-732
30. Kumar A.J., Köhler W., Kruse B. et al.; MR findings in adult-onset adrenoleukodystrophy; Am. J. Neuroradiol. 1995:16, 1227-1237
31. Kishimoto Y., Moser H.W., Kawamura N. et al.; Adrenoleukodystrophy: evidence that abnormal very long chain fatty acids of brain cholesterol esters are of exogenous origin; Biochem. Biophys. Res. Commun. 1980:96, 69-76
32. Rizzo W.B., Watkins P.A., Phillips M.W. et al.; Adrenoleukodystrophy: oleic acid lowers fibroblast saturated C22-26 fatty acids; Neurology 1986:36, 357-361
33. Bourre J.M., Daudu O., Baumann N. ; Nervonic acid biosynthesis by erucyl Co-A elongation in normal and quaking mouse brain microsomes. Elongation of other unsaturated fatty acyl-CoA (mono and polyunsaturated); Biochim. Biophys. Acta 1976:424, 1-7
34. Rizzo W.B., Leshner R.T., Odone A. et al.; Dietary erucic acid therapy for X-linked adrenoleukodystrophy; Neurology 1989:39, 1415-1422
35. Zierz S., Schröder R., Unkrig C.J.; Thrombocytopenia induced by erucic acid therapy in patients with X-linked adrenoleukodystrophy; Clin. Investig. 1993:71, 802-805
36. Poulos A., Gobson R., Sharp P. et al.; Very long chain fatty acids in X-linked adrenoleukodystrophy brain after treatment with Lorenzo’s oil; Ann. Neurol. 1994:36, 741-746
37. Van Geel B.M., Assies J., Haverkort E.B. et al.; Progression of abnormalities in adrenomyeloneuropathy and neurologically asymptomatic X-linked adrenoleukodystrophy despite treatment with ’’Lorenzo’s oil” ; J. Neurol. Neurosurg. Psychiatry 1999:67, 290-299
38. Moser H.W., Tutschka P.J., Brown III F.R. et al.; Bone marrow transplant in adrenoleukodystrophy; Neurology 1984:34, 1410-1417
39. Aubourg P., Blanche S., Jambaqué I. et al.; Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation; N. Engl. J. Med. 1990:322, 1860-1866
40. Krivit W., Lockman L.A., Watkins P.A. et al.; The future for treatment by bone marrow transplantation for adrenoleukodystrophy, metachromatic leukodystrophy, globoid cell leukodystrophy and Hurler syndrome; J. Inher. Metab. Dis. 1995:18, 398-412
41. Naidu S., Bresnan M.J., Griffin D. et al.; Childhood adrenoleukodystrophy. Failure of intensive immunosuppression to arrest neurological progression; Arch. Neurol. 1988:45, 846-848
42. Korenke G.C., Christen H., Hunneman D.H. et al.; Failure of β-IFN therapy in X-linked adrenoleukodystrophy; Eur. J. Pediatr. 1996:155, 833
43. Brown III F.R., van Duyn M.A., Moser A.B. et al.; Adrenoleukodystrophy: effects of dietary restriction and of administration of carnitine and clofibrate on clinical status and plasma fatty acids; Johns Hopkins Med. J. 1982:151, 164-172
44. Petroni A., Papini N., Blasevich M. et al.; Testosterone metabolites in patients reduce the levels of very long chain fatty acids accumulated in X-adrenoleukodystrophic fibroblasts; Neurosci. Letters 2000:289, 139-142
45. Cartier N., Lopez J., Moullier P. et al.; Retroviral-mediated gene transfer corrects very long chain fatty acid metabolism in adrenoleukodystrophy fibroblasts; Proc. Natl. Acad. Sci. USA 1995:92, 1674-1678
46. Chen S., Sawicka J., Betterle C. et al.; Autoantibodies to steroidogenic enzymes in autoimmune polyglandular syndrome, Addison’s disease and premature ovarian failure; J. Clin. Endocr. Metab. 1996:81, 1871-1876