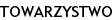Endokrynologia Pediatryczna, Tom 6 | Rok 2007 | Nr 1(18)
DOI: 10.18544/EP-01.06.01.0019
Aspekty zaburzeń stomatognatycznych u 6-letniego chłopca z zespołem Silvera-Russela
1Zakład Protetyki Stomatologicznej Akademii Medycznej w Białymstoku
2II Klinika Chorób Dzieci Dziecięcego Szpitala Klinicznego w Białymstoku
3NZOZ „Praktyka stomatologiczna M. Pacuk” w Wasilkowie
Słowa kluczowe: zespół Silvera-Russela, niski wzrost, klinodaktylia, mikrognacja, hipodoncja
Streszczenie
Zespół Silvera-Russela (ZSR) występujący z częstością od 1: 3 000 do 1: 100 000 urodzeń jest zespołem wad rozwojowych, któremu towarzyszy upośledzenie rozwoju wewnątrzmacicznego i wzrastania pourodzeniowego. Diagnozę opartą na analizie fenotypu (niski wzrost, mała masa urodzeniowa, asymetria różnych okolic ciała, zaburzenia rozwoju płciowego, charakterystyczny wygląd twarzoczaszki, plamy na skórze typu cafe-au-lait, klinodaktylia, syndaktylia) uzupełnia się między innymi o badania cytogenetyczne. Aktualnie nie ustalono jednoznacznie etiopatogenezy ZSR, choć udział czynników genetycznych staje się coraz bardziej znamienny. Przykładem jest identyfikacja u wielu pacjentów z ZSR matczynej duplikacji lub hipometylacji telomeru 11p15, disomii chromosomu 7, czy też stosunkowo często występujących abberacji chromosomów 8, 15, 17, 18 i 20. W obrazie klinicznym poza zasadniczymi wyznacznikami zespołu, częstym elementem są również zaburzenia układu stomatognatycznego o podłożu morfologicznym i czynnościowym. Głównie są to: mikrognacja, retrognacja, hipodoncja, mikrodoncja, stłoczenia zębów, zaburzenia ząbkowania oraz duża podatność na próchnicę. Prezentowany sześcioletni pacjent ma wiele cech charakterystycznych dla zespołu Silvera-Russela. Dodatkowo obciążony jest niedosłuchem przewodzeniowym i wykazuje upośledzenie rozwoju mowy. Ponadto, wskutek znacznej podatności na próchnicę i przy zaniedbaniu higieny, chłopiec utracił większość zębów mlecznych. Doprowadziło to do pogłębienia niektórych cech zespołu, utrudnienia prawidłowej diety dziecka i komplikacji terapii zaburzeń towarzyszących. Autorzy wskazują potrzebę wielospecjalistycznej opieki nad pacjentami z ZSR.
Wstęp
Zespół Silvera-Russela (ZSR) występujący z częstością od 1: 3 000 do 1: 100 000 urodzeń jest zespołem wad rozwojowych, któremu towarzyszy upośledzenie rozwoju wewnątrzmacicznego i wzrastania pourodzeniowego [1]. Po raz pierwszy został opisany w latach pięćdziesiątych ubiegłego stulecia. Przyczyny zespołu nie zostały jeszcze jednoznacznie zdefiniowane [2]. Początkowo przypisywano istotne znaczenie etiologiczne embriopatii ujawniającej się w 6-7 tygodniu życia płodowego. Obecnie ze względu na obserwowane różnego rodzaju dominujące mutacje ZSR został wpisany do rejestru MIM (Mendelian Inheritance in Man) jako zaburzenie autosomalne dominujące [3]. Dotychczas najczęstszą abberacją odkrytą u 10% pacjentów była disomia 7 chromosomu matczynego (mUPD7) oraz zaburzenia związane z chromosomem 17, natomiast aktualnie kluczową rolę w powstawaniu fenotypu ZSR przypisuje się dysregulacji genów chromosomu 11p15 w postaci matczynej duplikacji lub hipometylacji telomeru, a także hipometylacji genu H19 odpowiedzialnego za działanie czynnika wzrostu IGF 2 [4–10].
Do rozpoznania zespołu stosuje się opisane przez Lai i wsp. kryteria, które oparte są na najczęstszych objawach klinicznych [11]. Dokładny obraz zespołu jest bardzo trudny do uchwycenia ze względu na wielofenotypowość. Mimo licznych opisów, nie została sprecyzowana moc diagnostyczna poszczególnych objawów towarzyszących niskiemu wzrostowi [12, 13].
Niezmiernie ważnym elementem, mającym znaczenie zarówno przy diagnostyce, jak i przy terapii, jest zaburzenie kostnienia. U większości chorych stwierdza się charakterystyczny dla tego zespołu opóźniony wiek kostny względem wieku metrykalnego. Dyskrepancja ta ulega zmianom w trakcie rozwoju dziecka: początkowo wzrasta do około siódmego roku życia, a następnie stopniowo maleje do całkowitego zaniku około 12 roku życia [2, 12]. Istotnym objawem zespołu jest wygląd twarzoczaszki z nisko osadzonymi uszami, adekwatnym dla wieku obwodem głowy, wysoko zarysowaną linią włosów, wydatnie uwypuklonym czołem i mocno zaznaczonymi guzami oraz pomniejszonymi wymiarami reszty ciała, co daje wrażenie zbyt dużej czaszki (pseudohydrocephalus). Gałki oczne są głęboko osadzone, często obecne antymongoidalne ustawienie szpar powiekowych; sporadycznie występuje asymetria twarzy oraz opadnięcie powieki po stronie mniej rozwiniętej [2]. Nos mały, z nozdrzami skierowanymi ku przodowi. Usta wąskie, w kształcie odwróconej litery ‘V’ (tzw. shark’s mouth). Niedorozwinięta żuchwa często pozostaje cofnięta (micrognatia, retrognatia) [2]. Przeprowadzone dokładne badanie stomatologiczne ujawnia wady zgryzu (zazwyczaj uwarunkowane gnatycznie), stłoczenia zębów, gotyckie podniebienie, opóźnione ząbkowanie wraz z zaburzeniem kolejności wyrzynania, mikrodoncją oraz hipodoncją [13, 14, 15]. Ponadto znaczną podatność na próchnicę u tych chorych stwierdza się oraz skłonność do przedwczesnej utraty zębów. Rzadziej występują różnice w szerokości zębów prawej i lewej połowy twarzy – u osób z asymetryczną budową ciała występują mniejsze wymiary mezjalno-dystalne po stronie skróconych kończyn. Dodatkowo obserwuje się odmienną morfologię radiologiczną zębów jednoimiennych danego łuku [13].
Poza wcześniej wspomnianymi kryteriami, na uwagę zasługują następujące komponenty: szczupła budowa ciała (niedobór tkanki tłuszczowej oraz mięśniowej), zaburzenia szkieletowe (skolioza, lordoza, wklęsła klatka piersiowa, deformacje kończyn i stawów biodrowych), wady serca, powiększenie narządów miąższowych (wątroby, śledziony, nerek), defekty układu moczowo-płciowego, zaburzenia hormonalne, nadmierna potliwość, wysoki, piskliwy głos, ubytek słuchu [2, 12]. Chorzy z ZSR znajdują się w grupie o zwiększonym prawdopodobieństwie nowotworów [1].
Znaczny odsetek pacjentów wykazuje deficyty funkcji poznawczych oraz mowy; nie należy do rzadkości opóźniony rozwój psychomotoryczny, zaś ok. 15% przypadków jest upośledzonych umysłowo [1]. Obniżona samoocena i problemy emocjonalne związane z wyglądem to powszechne rysy w psychice starszych pacjentów.
Opis przypadku
Chłopiec J.S. (ryc. 1a, 1b, 1c) (nr hist. chor.: 3967/2005) urodzony z ciąży III prawidłowej i porodu III prawidłowego, siłami natury w 36 tyg. ciąży, z masą urodzeniową 1550, długością ciała 25,4 cm i 4 punktami w skali Apgar, z rozpoznanym zespołem Silvera-Russela został skierowany do II Kliniki Chorób Dzieci AMB z Poradni Genetycznej SPDSK w Białymstoku celem poszerzenia diagnostyki endokrynologicznej. Niedobór masy ciała i wzrostu oraz asymetria twarzy były obserwowane od urodzenia. W okresie noworodkowym i niemowlęcym dziecko często chorowało na infekcje górnych i dolnych dróg oddechowych. Rodzice chłopca i jego rodzeństwo są zdrowe, średniego wzrostu (ojciec i brat 25-50 p. centyl, matka i siostra 50 p. centyl), z prawidłowym przebiegiem pokwitania. Wywiad rodzinny w kierunku zaburzeń szkieletowych, endokrynologicznych (w tym zaburzeń wzrastania) i zespołów wad rozwojowych był ujemny.
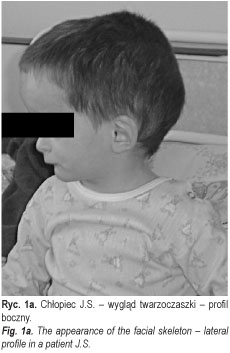
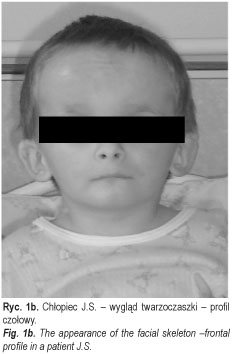
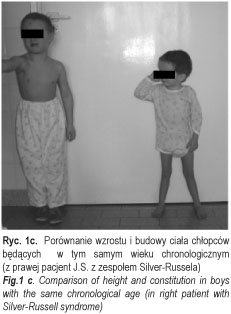
W chwili przyjęcia do Kliniki wiek chronologiczny chłopca wynosił 5 8/12 lat. W badaniu fizykalnym stwierdzono znaczny niedobór wzrostu (-7SDS) i masy ciała (znacznie < 3 p. centyla), słabo rozwiniętą tkankę mięśniową, przedpokwitaniowy rozwój narządów płciowych, prącie długości 1,5 cm, objętość obu jąder 1/2 ml (według orchidometru Pradera) okresowo przemieszczających się do kanału pachwinowego (typ wędrujący). W budowie ciała istotnie zwracała uwagę mała, trójkątna asymetryczna twarz, nisko osadzone uszy, wydatnie uwypuklone czoło z mocno zaznaczonymi guzami czołowymi, drobne dłonie i stopy, klinodaktylia 5-go palca dłoni, syndaktylia 2-go i 3-go palca stóp, koślawość kończyn dolnych oraz asymetryczna budowa tułowia. Wykonane analizy hormonalne ujawniły nieco podwyższony poziom TSH, istotnie obniżone stężenie IGF-1, przedpokwitaniowe wartości hormonów płciowych oraz prawidłowy profil kortyzolu. Badania oceniające profil nocny GH po zaśnięciu i testy rezerw przysadkowych GH po Glukagonie (0,03 mg/kg i.v.) i L-dopie (300mg/m2 po) wykazały we wszystkich próbach obniżony poziom hormonu wzrostu. Rtg PA lewej dłoni uwidoczniło nierównomierny rozwój kości ręki; wiek kości nadgarstka oceniono na ok. 2 lata, zaś śródręcza i palców – na ok. 3 lata. Badanie CT przysadki mózgowej wykluczyło zmiany ogniskowe. W badaniu USG jamy brzusznej stwierdzono powiększenie prawego płata wątroby. Badanie elektrokardiograficzne wykazało zaburzenia przewodnictwa w postaci bloku prawej odnogi pęczka Hisa, zaś echokardiograficzne serca – poszerzoną przegrodę międzykomorową serca. W konsultacji logopedycznej oceniono sprawność i kompetencję językową jako opóźnioną (dziecko stosowało wyrazy strzępkowe, przestawki agramatyczne, przestawiało głoski i sylaby, używało mowy dziecinnej, nie zmiękczało części głosek, nosowało). Przeprowadzona konsultacja laryngologiczna wykryła niedosłuch przewodzeniowy. W konsultacji okulistycznej nie stwierdzono szczególnych zaburzeń, poza podejrzeniem proliferacji gleju na tarczach nerwu wzrokowego; ostrości wzroku nie można było ocenić.
Wykonane szczegółowe badanie stomatologiczne zewnątrzustne wykazało mikrognację, z dysproporcją obu szczęk (żuchwa znacznie mniej rozwinięta). Okolica podnosowa zapadnięta, profil skośny do tyłu, proporcje twarzy znacznie zaburzone (dominacja górnego odcinka, środkowe i dolne piętro – mocno pomniejszone). Wewnątrzustnie stwierdzono bezzębie szczęki (pacjent utracił większość zębów mlecznych w trzecim roku życia) oraz resztkowe uzębienie mleczne w żuchwie w postaci drugich trzonowców (z dużymi ubytkami) i mocno zniszczonych zębów siecznych. Wyrostki zębodołowe szczęki są dobrze ukształtowane na całym obwodzie, ale nie wykazują oznak rychłego wyrzynania się zębów stałych; podniebienie jest średnio wysklepione. Przeprowadzone badanie radiologiczne pod postacią pantomogramu (ryc. 1) wykazało następujące zaburzenia: brak zawiązków zębów przedtrzonowych szczęki strony lewej, brak zawiązków drugich zębów przedtrzonowych żuchwy obu stron, zniekształcony zawiązek zęba 44. Ze względu na słabą ostrość zdjęcia w górnym środkowym sekstancie nie można jednoznacznie stwierdzić nieobecności zawiązków zębów 13 i 12. W obu szczękach widoczna jest skąpa ilość substancji kostnej o delikatnym ubeleczkowaniu i wręcz ażurowej strukturze – zawiązki zębów zdają się stanowić większą część objętości kości.
Aktualne leczenie stomatologiczne pacjenta opiera się jedynie na leczeniu zachowawczym pozostałego uzębienia mlecznego. Leczenie protetyczne, jakkolwiek wskazane ze względu na całkowity brak zębów mlecznych w szczęce, nie jest możliwe do przeprowadzenia ze względu na skrajnie negatywną postawę dziecka. W leczeniu zaś hormonalnym wprowadzono L-tyroksynę doprowadzając do normalizacji poziomu tyreotropiny oraz przeprowadzono 5-tygodniową terapię Biogonadylem, uzyskując efekt sprowadzenia obu jąder do worka mosznowego. W chwili obecnej pacjent znajduje się pod wielospecjalistyczną opieką.
Dyskusja
Pacjenci z zaburzeniami zespołowymi powinni być bez wątpienia poddawani terapii wielospecjalistycznej, pozwalającej na optymalne dostosowanie jej do etapu rozwoju dziecka [1]. Biorąc pod uwagę, iż głównym wyznacznikiem, a zarazem i bolączką pacjentów z zespołem Silvera-Russela jest niski wzrost – duże znaczenie mają tu działania mające na celu umożliwienie pacjentowi osiągnięcie potencjalnie najwyższego wzrostu. Możliwe jest leczenie z użyciem rekombinowanego hormonu wzrostu, ale osiągane efekty są słabiej wyrażone niż u dzieci w innych zbliżonych grupach z IUGR [1, 2, 16]. Ważną rolę ma właściwie dobrana wysokokaloryczna dieta. W przypadku asymetrii budowy ciała wskazana jest korekta ortopedyczna, zaś w skrajnych przypadkach – interwencja chirurgiczna [11]. Nierzadko interwencji terapeutycznej wymaga zaburzone pokwitanie. Konieczna jest kompleksowa opieka i rehabilitacja neurologiczna, logopedyczna i psychologiczna.
Chociaż z wiekiem charakterystyczny obraz twarzy pacjentów z zespołem Silvera-Russela ulega zatarciu, to leczenie zaburzeń w obrębie twarzoczaszki nie może być traktowane marginalnie [1]. Od początku, po stwierdzeniu nieprawidłowości, dziecko powinno znaleźć się pod czynną opieką lekarza stomatologa, najlepiej specjalisty ortodonty [14]. W przypadku znacznych, utrzymujących się po osiągnięciu stabilizacji wzrostowej dysproporcji/deformacji twarzoczaszki (uwzględniając również wpływ terapii hormonalnej), należy wziąć pod uwagę możliwość leczenia chirurgicznego pod postacią osteotomii korekcyjnej z użyciem dystraktorów kostnych i/lub przeszczepów autogennych, a także zespoleń mikro- lub minipłytkami. Zależnie od postaci morfologicznej zniekształcenia rozległość zabiegu obejmuje jedną lub obie kości szczęk, kości jarzmowe, oczodoły i kości nosa. Najczęściej w zaburzeniach dysmorficznych twarzoczaszki stosowane są osteotomie typu Le Fort I lub Le Fort II, z następczym wysunięciem szczęk ku przodowi. Plan osteotomii korekcyjnej musi być opracowany szczegółowo na podstawie analizy modeli gipsowych i pomiarów kraniometrycznych, we współpracy wielospecjalistycznej. Obowiązuje dokładne ustalenie linii przecięć, sposobu unieruchomienia odłamów, rozważenie przeszczepu kości oraz miejsca jego pobrania. W postępowaniu przed- jak i pooperacyjnym konieczna jest niekiedy długotrwała rehabilitacja ortopedyczna i psychologiczna oraz leczenie protetyczne chorego [17, 18].
W przypadku hipodoncji uzasadnionym wydaje się włączenie do zespołu terapeutycznego lekarza protetyka. Wspólna opieka ortodontyczno-protetyczna stanowi ważną składową w prowadzeniu pacjenta w takiej sytuacji [19]. Pacjent J.S. nie posiada zawiązków zębów 24, 25, 35, 45, zaś pod znakiem zapytania pozostaje obecność zawiązków zębów 13 i 12. Brak drugich przedtrzonowców żuchwy będzie mógł zostać łatwo skompensowany odpowiednim ustawieniem pozostałych zębów (w przypadku korzystnego wyniku badań kraniometrycznych), natomiast odpowiednia korekcja w szczęce będzie sprawiała więcej problemów. Wskazane jest wykonanie protezy dziecięcej lub aparatu ortodontycznego tak szybko, jak tylko pozwoli na to ilość wyrżniętych zębów oraz kondycja psychofizyczna pacjenta [18, 19].
Nie bez znaczenia pozostaje również znaczna podatność na próchnicę (także uzębienia mlecznego) chorych z ZSR. Pacjent J.S. w wyniku zaawansowanej choroby próchnicowej i związanych z nią powikłań utracił już w trzecim roku życia ponad połowę zębów mlecznych (wg dokumentacji medycznej: z przyczyn doraźnych dokonano ekstrakcji zębów 62, 63, 64 i 74, natomiast korzenie zębów 54, 53, 52, 51, 61, 73, 83, 84 zostały usunięte w znieczuleniu ogólnym). Obecny stan bezzębia szczęki i uzębienia resztkowego żuchwy z całą pewnością nie pomaga w utrzymywaniu odpowiedniej diety dziecka oraz kształtowaniu wymowy. Bezdyskusyjny pozostaje fakt braku bodźcowania ze strony aktywnych zębów na układ stomatognatyczny oraz utrzymywania/kształtowania przestrzeni dla zębów stałych [14, 15]. Dlatego też niezmiernie istotne jest dokładne poinformowanie rodziców chorego dziecka o konieczności częstych wizyt kontrolnych w specjalistycznym gabinecie stomatologicznym oraz przede wszystkim utrzymywania wzorowej higieny jamy ustnej małego pacjenta.
Podsumowując, należy podkreślić, iż w obrazie klinicznym zespołu Silvera-Russela istotną rolę odgrywają zaburzenia stomatognatyczne, których wczesna, właściwa diagnostyka i konsekwentne leczenie oparte na specjalistycznej opiece ortodontyczno-protetycznej może poprawić komfort życia pacjenta i zapobiec niekorzystnym następstwom opisywanych zaburzeń.
Piśmiennictwo
1. Prakash-Cheng A., McGovern M.; Silver-Russell Syndrome; eMedicine Spec. Ped. Gen. & Met. Dis. 2003:12, 1 [http://www.emedicine.com/ped/topic2099.htm]
2. Price S.M., Stanhope R., Garrett C. et al.; The spectrum of Silver-Russell syndrome: a clinical and molecular genetic study and new diagnostic criteria; J. Med. Genet. 1999:36, 837-42
3. Ounap K., Reimand T., Magi M.L. et al.; Two sisters with Silver-Russell phenotype; Am. J. Med. Genet. A. 2004:3, 301-6
4. Ayala-Madrigal M.L., Shaffer L.G., Ramirez-Duenas M.L.; Silver-Russell syndrome and exclusion of uniparental disomy; Clin. Genet. 1996:6, 494-7
5. Hitchins M.P., Stanier P., Preece M.A., et al.; Silver-Russell syndrome: a dissection of the genetic aetiology and candidate chromosomal regions; J. Med. Genet. 2001:12, 810-9
6. Schonherr N., Meyer E., Eggermann K. et al.; (Epi)mutations in 11p15 significantly contribute to Silver-Russell syndrome: but are they generally involved in growth retardation?; Eur. J. Med. Genet. 2006:29
7. Gicqel C., Rossignol S., Cabrol S. et al.; Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome; Nat. Genet. 2005:9, 1003-7
8. Bliek J., Terhal P., van den Bogaard M.J. et al.; Hypomethylation of the H19 gene causes not only Silver-Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype; Am. J. Hum. Genet. 2006:4, 604-14
9. Eggermann T., Meyer E., Ranke M.B. et al.; Diagnostic proceeding in Silver-Russell syndrome; Mol. Diagn. 2005:4, 205-9
10. Flori E., Girodon E., Samama B. et al.; Trisomy 7 mosaicism, maternal uniparental heterodisomy 7 and Hirschsprung’s disease in a child with Silver-Russell syndrome; Eur. J. Hum. Genet. 2005:9, 1013-8
11. Lai K.Y.C., Skuse D., Stanhope R., Hindmarsh P.; Cognitive abilities associated with the Silver-Russell syndrome; Arch. Dis. Child. 1994:71, 490-496
12. Abraham E., Altiok H., Lubicky J.P.; Musculosceletal manifestations of Russell-Silver Syndrome; J. Pediatr. Orthop. 2004:24, 552-564
13. Bedi R., Moody G.H.; A primary double molar tooth in a child with Russell-Silver syndrome; Br. Dent. J. 1991:9, 284-6.
14. Cullen C.L., Wesley R.K.; Russell-Silver syndrome: microdontia and other pertinent oral findings; ASDC J. Dent. Child. 1987:3, 201-4
15. Azcona C., Stanhope R.; Absence of catch-down growth in Russell-Silver syndrome after short-term growth hormone treatment; Horm. Res. 1999:1, 47-9
16. Bartkowski S.B.; Chirurgia szczękowo-twarzowa; Oficyna Wyd. „AGES” Kraków 1996, 405-6
17. Karłowska I.; Zarys współczesnej ortodoncji; Wyd. Lek. PZWL Warszawa 2001, 312-327
18. Spiechowicz E.; Protetyka stomatologiczna; Wyd. Lek. PZWL Warszawa 1998, 532-537