Endokrynol. Ped. 11/2012;3(40):81-88
DOI: 10.18544/EP-01.11.03.1393
Zespół Prader-Willi – diagnostyka i leczenie
1Klinika Endokrynologii i Diabetologii Dziecięcej, Uniwersytet Medyczny w Lublinie
2Klinika Patologii Noworodków Niemowląt, Uniwersytet Medyczny w Lublinie
Słowa kluczowe: zespół Prader-Willi
Streszczenie
Zespół Prader-Willi to zespół wad wrodzonych, występujący z częstością 1:10000–1:30000 żywo urodzonych, uwarunkowany genetycznie. Spowodowany jest nieprawidłowością w regionie 15 pary chromosomu (15q11–13) określanego jako region krytyczny zespołu Prader-Willi. Opis przypadku. Dziewczynka z CIPI, wykazująca słabe ruchy płodu w okresie ciąży, u której obserwowano trudności ze ssaniem i hipotrofię w okresie noworodkowym, a następnie opóźnienie rozwoju psychoruchowego i hypertermię w okresie niemowlęcym była diagnozowana w kierunku zespołu Prader-Willi i rozpoznano u niej delecję fragmentu ojcowskiego chromosomu 15q11.2–q13. Pacjentka jest leczona
w sposób zintegrowany: otrzymuje ludzki rekombinowany hormon wzrostu, uczestniczy w zajęciach rehabilitacyjnych, ma wdrożoną dietę niskokaloryczną, uczestniczy w zajęciach z psychologiem i logopedą. Stosowane leczenie umożliwia prawidłowy rozwój fizyczny dziecka i optymalny rozwój umysłowy
Wstęp
Zespół Prader-Willi (PWS) to zespół wad wrodzonych, występujący z częstością 1:10000–1:30000 żywo urodzonych, uwarunkowany genetycznie. Spowodowany jest nieprawidłowością w regionie 15 pary chromosomu (15q11–13), który jest określany jako region krytyczny zespołu Prader-Willi, powstałą na skutek jednego z kilku mechanizmów: delecji fragmentu ojcowskiego chromosomu 15q11.2–q13 (około 70%), uniparentnej (jednorodzicielskiej – matczynej) heterodisomii (UPD, uniparental disomy) chromosmomu 15, mutacji imprintingowej (mikrodelecji w miejscu imprintingowym) przekazanej na chromosomie ojcowskim, zrównoważonej rearanżacji chromosomalnej w obrębie 15q11.2–q13 (mniej niż 1%) [1,2].
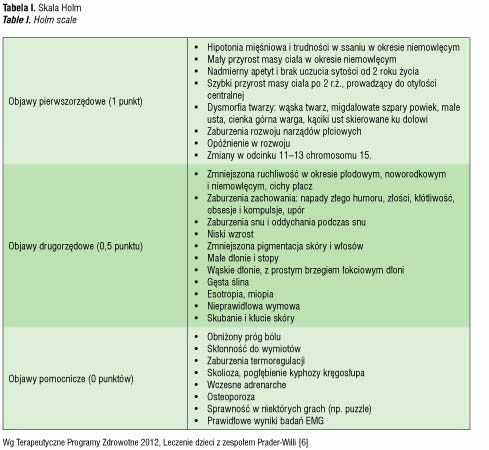
W zależności od wieku obserwuje się różną manifestację cech klinicznych zespołu. Jest to często przyczyną utrudniającą rozpoznanie, w szczególności u najmłodszych pacjentów. Do rozpoznania klinicznego zespołu użyteczna jest skala wg Holm [3,4], która dzieli objawy na pierwszo- i drugorzędowe oraz pomocnicze. Objawy są punktowane: 1 punkt dla objawów pierwszorzędowych, 0,5 dla drugorzędowych i 0 punktów dla objawów pomocniczych (tab. I). PWS można rozpoznać u:
• dzieci <3r.ż. z sumą punktów minimum5,
• dzieci >3r.ż. z sumą punktów minimum 8.
Potwierdzeniem rozpoznania jest badanie cytogenetyczne – stwierdzenie delecji długiego ramienia chromosomu 15 pochodzenia ojcowskiego lub disomii matczynej chromosomu 15.
Opis przypadku
Opisywana dziewczynka, urodzona z ciąży pierwszej, porodu pierwszego w 35 hbd, rozwiązanego przez cięcie cesarskie w zielonych wodach płodowych, masa ur. 2100 g, ocena w skali Apgar 6/7/9 punktów, została przyjęta do Kliniki Patologii Niemowląt w wieku 11 miesięcy z powodu stanów gorączkowych utrzymujących się od 2 tygodni bez klinicznych i laboratoryjnych wykładników infekcji. W okresie okołoporodowym przebywała w Oddziale Intensywnej Terapii z powodu wrodzonej posocznicy, niewydolności wielonarządowej, trombocytopenii oraz zapalenia płuc przebiegającego z niewydolnością oddechową. Od urodzenia stwierdzano obniżone napięcie mięśniowe, trudności w karmieniu (zaburzenia ssania i połykania) oraz osłabione odruchy ścięgniste. W okresie niemowlęcym dziecko 4-krotnie hospitalizowano z powodu nawracających infekcji dróg oddechowych, zapalenia płuc, wiotkości mięśni, opóźnienia psychomotorycznego oraz pojedynczego epizodu drgawek. W przeprowadzonej diagnostyce wykluczono toksoplazmozę, cytomegalię, różyczkę, choroby metaboliczne, miopatię, rdzeniowy zanik mięśni oraz niedoczynność tarczycy. W Klinice Patologii Noworodków rozpoznano niedorozwój ciała modzelowatego, zaniki korowo-podkorowe, szeroki zbiornik rdzeniowo-móżdżkowy.
W dniu przyjęcia do Kliniki Patologii Niemowląt stan ogólny dziecka oceniono jako dobry, dziecko było wydolne krążeniowo i oddechowo. W badaniu fizykalnym stwierdzono opóźnienie rozwoju psychomotorycznego (dziecko nie siedziało samodzielnie), obniżoną aktywność ruchową, znacznego stopnia hipotonię mięśniową, otyłość, słabe wykształcenie warg sromowych większych, owalną twarz, migdałowate oczy, ubogą mimikę, nisko osadzone małżowiny uszne, wysokie gotyckie podniebienie. W wykonanych podstawowych badaniach laboratoryjnych nie stwierdzono odchyleń. W zdjęciu rtg klatki piersiowej opisywano sylwetkę serca nieco powiększoną, rysunek naczyniowy płuc obfity, linijne zacienienie obwodowo po stronie płuca prawego – zwłóknienie opłucnowe. Na podstawie wywiadu, badania przedmiotowego, badań dodatkowych, obserwacji klinicznej oraz konsultacji (neurologicznej, psychologicznej i rehabilitacyjnej) wysunięto podejrzenie zespołu Pradera-Williego. Utrzymywanie się stanów podgorączkowych i gorączki wiązano z zaburzeniami termoregulacji w obrębie podwzgórza. Zdecydowano się na przeprowadzenie badania genetycznego, w którym stwierdzono delecję ramienia długiego chromosomu 15 (15q11–13). Na tej podstawie postawiono rozpoznanie zespołu Pradera-Willego.
Fenotyp dziecka był charakterystyczny dla PWS w okresie poniemowlęcym, jak i później. Szczególną uwagę zwracały typowa dysmorfia twarzy: pociągła twarz z wąskim wymiarem dwuskroniowym, kształt ust (wąska górna warga, kąciki zwrócone ku dołowi), oczy (migdałowaty kształt, hipoteloryzm, zmarszczka nakątna). Dodatkowe cechy budowy ciała : niskorosłość, małe dłonie i stopy, jasna karnacja skóry i włosów także włączały się w obraz zespołu.
Dziewczynka w wieku 2,5 roku została zakwalifikowana do programu leczenia hormonem wzrostu ze względu na niskorosłość jako składową zespołu PWS. Od tamtej pory pozostaje pod regularną wielospecjalistyczną kontrolą, w tym endokrynologiczną. Rozwój psychoruchowy dziewczynki w okresie poniemowlęcym przebiegał z opóźnieniem. Samodzielnie siadać zaczęła w wieku 1 roku i 4 miesięcy, pierwsze kroki postawiła rok później, zaś gdy miała 2,5 roku, chodziła przy pomocy podtrzymywania za ręce, samodzielnie po kolejnych 3–4 miesiącach. Już po pierwszych 3 miesiącach leczenia ludzkim rekombinowanym hormonem wzrostu (HGH) obserwowano stopniową poprawę napięcia mięśniowego dziewczynki, co przekładało się na coraz sprawniejsze poruszanie się i ogólne zwiększenie aktywności ruchowej.
Początkowe problemy z karmieniem dziecka w okresie niemowlęcym ustąpiły po ukończeniu 1 r. ż., od 2 r.ż. rodzice zauważyli zwiększanie się apetytu aż do nadmiernego w 3 r.ż. Masa ciała dziewczynki przed włączeniem terapii HGH (1rok i 7 mies.) była pomiędzy 10–25 centylem, zaś na siatce proporcji masy do wysokości ciała pomiary mieściły się w przedziale 50–75centyla. Rodziców przeszkolono w zakresie diety niskokalorycznej i sposobu karmienia dziecka, uwzględniając ciągłe uczucie głodu w związku z uszkodzeniem ośrodka sytości w podwzgórzu oraz mniejsze zapotrzebowanie energetyczne u pacjentów z zespołem Prader-Willi. Po 8 miesiącach, nadal jeszcze przed włączeniem terapii HGH, masa ciała była w przedziale 3–10 centyla, a wskaźnik proporcji masy ciała do wysokości w granicach 50–75centyla. Dzięki ogromnemu zaangażowaniu i determinacji rodziców dziewczynka, pomimo rozwijającego się coraz bardziej typowego dla jej choroby nieustającego uczucia głodu, dotychczas nie rozwinęła otyłości (ryc. 1, 2). Obecna dieta dziecka zawiera 1000 kcal/dobę, co stanowi 75% zapotrzebowania energetycznego dla jego wieku z rozdziałem: I śniadanie 250 kcal, II śniadanie 150 kcal, obiad 250 kcal, podwieczorek 150 kcal, kolacja 200 kcal. O konieczności zachowania rygorystycznej diety poinformowane są wszystkie osoby mające kontakt z dzieckiem – członkowie rodziny oraz pracownicy przedszkola.
Poza terapią HGH oraz stosowaną dietą w prawidłowym rozwoju pacjentki i zapobieganiu otyłości niezmiernie ważna jest codzienna aktywność ruchowa, jest to trzecia składowa tzw. terapii zintegrowanej. Mimo że – porównując do typowych dzieci z PWS – dziewczynka jest dość aktywna na co dzień, rodzice podają, że chętniej wybiera jednak zajęcia i zabawy stacjonarne niż ruchowe, potrzebuje specjalnej zachęty i motywacji. Zgodnie z zaleceniami dla pacjentów z PWS rodzice stworzyli dla niej ściśle określony i pilnie przestrzegany plan dnia z wpisanymi regularnymi posiłkami oraz czasem przeznaczonym na aktywność ruchową – ćwiczenia fizyczne wykonywane wspólnie 3–4 razy w ciągu dnia przed posiłkami (co jest dodatkową motywacją – dziecko wie, że po ćwiczeniach dostanie coś do jedzenia). Część zajęć fizycznych odbywa się w przedszkolu. Dodatkowo dziewczynka uczęszcza na zajęcia rehabilitacyjne kilka razy w tygodniu.
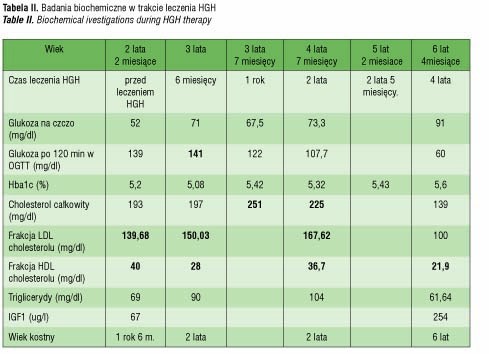
Dzięki przestrzeganiu trzech powyższych zasad terapii zintegrowanej : leczenia HGH, stosowania diety o obniżonej kaloryczności oraz stałego programu aktywności fizycznej dziewczynka osiąga prawidłowe dla wieku parametry rozwoju fizycznego (masa ciała, wzrost i BMI 50 centyl). Nie rozwinęła także metabolicznych powikłań ani w przebiegu zespołu, ani wskutek ewentualnych działań niepożądanych HGH. Obserwowano jedynie przejściowe zaburzenia gospodarki węglowodanowej po 6 miesiącach leczenia (graniczny poziom glikemii w 120 min testu OGTT przy prawidłowej wartości hemoglobiny glikowanej), które nie powtórzyły się w kolejnych badaniach kontrolnych, oraz zaburzenia gospodarki lipidowej zarówno przed, jak i przez pierwsze dwa lata terapii HGH. Początkowo znacznie opóźniony wiek kostny w 6 r.ż. odpowiada chronologicznemu. Porównanie podstawowych badań biochemicznych, hormonalnych i ocenę wieku kostnego przed włączeniem i w trakcie terapii HGH obrazuje tabela 2.
Podczas diagnostyki poprzedzającej leczenie GH stwierdzono u dziewczynki niedoczynność tarczycy, w związku z czym włączono leczenie substytucyjne lewo tyroksyną, zapewniające eutyreozę.
W trakcie terapii HGH dziewczynka podlega kontroli laryngologicznej, kardiologicznej oraz ortopedycznej.
Badania laryngologiczne, włącznie z wideofiberoskopią jamy nosowo-gardłowej nie wykazały nieprawidłowości zarówno przed rozpoczęciem leczenia GH, jak i w czasie jego trwania.
U dziewczynki w okresie noworodkowym stwierdzono obecność PDA (którego nie uwidoczniono w kolejnych badaniach kontrolnych) oraz ślad IM, fizjologiczną IT. W 6 r.ż. – podobnie, jednak IM określono jako II stopnia, wymagającą kontroli za 6–12 miesięcy. W wykonanym w tym
samym czasie badaniu EKG nie stwierdzono patologii, cech przerostu ścian serca, jedynym odchyleniem od normy były dyskretne zaburzenia przewodnictwa śródkomorowego nad RV.
Podczas konsultacji ortopedycznej rozpoznano niewielką wadę postawy – zalecono wzbogacenie zajęć rehabilitacyjnych o ćwiczenie obręczy barkowej. W trakcie badań kontrolnych w 6 r.ż.
u dziewczynki zauważono cechy przedwczesnego dojrzewania w postaci rozwoju owłosienia łonowego przy braku rozwoju gruczołów piersiowych i owłosienia pachowego. Podwyższony poziom 17-OH progesteronu (7,30 ng/ml) przy prawidłowym poziomie przysadkowych i jajnikowych hormonów płciowych (FSH 7,35 U/l, LH <0,10 U/l, PRL 116,8 mU/l, Estradiol <18,4 pmol/l, Progesteron 1,01 nmol/l) wskazuje na nadnerczowe pochodzenie tych objawów, co jest charakterystyczne dla pacjentów z PWS.
Poważnym problemem, z jakim musi się zmagać pacjent z PWS i jego rodzina, są zaburzenia zachowania oraz opóźnienie w rozwoju umysłowym. Dotyczy to także opisywanej przez nas pacjentki. W badaniu psychologicznym skalą rozwoju psychomotorycznego, wykonanym gdy dziewczynka miała 1,5 roku, uzyskała ona iloraz rozwojowy globalny = 60, co odpowiadało ogólnemu poziomowi rozwoju psychomotorycznego dla dziecka 10-miesięcznego. W tym poszczególne sfery rozwojowe oceniono następująco: postawa i lokomocja – 8 mc, koordynacja wzrokowo-ruchowa – 10 mc, mowa – 9 mc, reakcje społeczne – 10 mc. Z wyniesionych sfer najsłabszą stroną badanej była lokomocja, a najmocniejszą uspołecznienie. W związku z powyższym dziewczynka została zakwalifikowana do stymulowania rozwoju z pomocą regularnej opieki psychologa i logopedy. Organizacyjnym ułatwieniem dla rodziców dziecka było korzystanie z pomocy przedszkola integracyjnego oraz poradni łączącej w jednym miejscu zabiegi rehabilitacyjne jak również porady psychologa i logopedy. Dzięki skutecznej współpracy tych specjalistów oraz bardzo odpowiedzialnej, pełnej zaangażowania i poświęcenia, postawie rodziców znacznie poprawił się rozwój intelektualny dziewczynki – w wieku 5 lat osiągnęła iloraz inteligencji = 82 (badanie z zastosowaniem Skali L. Termana i M. Merrill), co świadczy o normie intelektualnej mieszczącej się w obszarze poniżej przeciętnej. Poziom inteligencji płynnej badany Międzynarodową Wykonaniową Skalą Leitera znajdował się w obszarze normy (w granicy wyników poniżej przeciętnej do przeciętnych), iloraz dojrzałości społecznej oceniono jako znajdujący się w normie (82). W badaniu psychologicznym wykonanym w 6 r.ż. ogólny poziom rozwoju umysłowego oceniono na niższy niż przeciętny (szczególnie zauważalne wolne tempo pracy, rozproszona uwaga), odroczono spełnianie obowiązku szkolnego na rok.
Dziewczynka poczyniła postępy w rozwoju mowy: w 4 roku życia wypowiadała pojedyncze wyrazy, w 5 r.ż. zaczęła budować zdania, obecnie mówi płynnie, jednak nadal dość niewyraźnie. Spory problem stwarza labilność emocjonalna dziewczynki, łatwe wpadanie w złość, nieraz nawet w agresję i histerię, co utrudnia jej kontakty społeczne z rówieśnikami. Stałe uczucie głodu i niepokój z tym związany, a także tendencję do charakterystycznego skubania skóry i rozdrapywania drobnych ranek opiekunowie starają się wypełnić dziewczynce licznymi zajęciami angażującymi ją ruchowo lub umysłowo. Dziewczynka chętnie poddaje się rutynowemu, ustalonemu ściśle planowi dnia, nie lubi nagłych zmian w jego przebiegu.
Dyskusja
Pacjent z zespołem Pradera-Willego zwykle od pierwszych dni życia prezentuje problemy zdrowotne i rozwojowe, jednak wczesne postawienie właściwej diagnozy niejednokrotnie napotyka na problemy, spowodowane prawdopodobnie z jednej strony rzadkością występowania zespołu, z drugiej – różnorodnością nasilenia dysmorfii oraz cech charakterystycznych choroby [5]. Znaczną pomocą w usystematyzowaniu objawów i ustaleniu rozpoznania na podstawie objawów klinicznych jest kwestionariusz ustalony przez Holm i współpracowników [5,6]. Na jego podstawie opisana pacjentka w wieku 6 lat uzyskuje 11 punktów (na 8 koniecznych), w tym 7 z listy objawów pierwszorzędowych. U dziewczynki chorobę rozpoznano dużo wcześniej, w okresie niemowlęcym, natomiast kolejne charakterystyczne objawy przybywały z wiekiem. U pacjentów z PWS mogą wystąpić zaburzenia termoregulacji – tendencja do szybkiego wychładzania, ale też i do nagłych wzrostów temperatury, nawet bez udziału czynnika infekcyjnego, co ma źródło w uszkodzeniu ośrodka termoregulacji w podwzgórzu [5,6]. Takie zjawisko miało miejsce u opisywanej pacjentki w 11 mies. życia i łącznie z pozostałymi, bardziej charakterystycznymi, objawami klinicznymi dla PWS przyczyniło się do rozpoznania u niej choroby, potwierdzonej wynikiem badania genetycznego. Obecnie u dziewczynki nie obserwuje się niewyjaśnionych nagłych wzrostów temperatury, jednak – jak podają rodzice – ma ona tendencję do opóźnionej normalizacji temperatury ciała w sytuacjach przegrzania lub wychłodzenia.
Podstawowymi przyczynami problemów zdrowotnych osób z PWS są otyłość, niskorosłość, mała aktywność ruchowa i zaburzenia zachowania, napędzające wzajemnie błędne koło i w razie braku odpowiedniego leczenia nieuchronnie prowadzące do powstania patologicznej otyłości [5–8].
Otyłość w zespole PWS ma złożoną etiologię. Za podstawową przyczynę uważa się nieustające uczucie głodu, wynikające z uszkodzenia ośrodka sytości w podwzgórzu, dodatkowy wpływ mają zmniejszone zapotrzebowanie energetyczne (do 50% w porównaniu z osobami zdrowymi) oraz mniejsza aktywność ruchowa [5–8]. Na te cechy nakłada się dodatkowo niedobór hormonu wzrostu, który powoduje oprócz niskorosłości także zaburzenia metaboliczne i niedobór masy mięśniowej, pogłębiające niedobór aktywności ruchowej pacjentów [5–8]. Cechą charakterystyczną dzieci z PWS jest także podwyższone stężenie ghreliny, stymulującej łaknienie nawet już w pierwszych dwu latach życia przed wystąpieniem otyłości [7,9]. Ze względu na złożoność przyczyn otyłości w jej leczeniu nie wystarcza tylko stosowanie diety – podstawą jest tzw. terapia zintegrowana, polegająca na równoczesnym stosowaniu diety niskokalorycznej, terapii hormonem wzrostu w połączeniu z programem codziennej aktywności ruchowej [8]. Rygorystyczne przestrzeganie niskokalorycznej diety u pacjenta z nieustającym uczuciem głodu oraz dodatkowo z różnego stopnia upośledzonym rozwojem intelektualnym jest niezwykle trudne. Należy podkreślić, że nie można wymagać samodyscypliny od pacjenta, gdyż sam nigdy nie będzie w stanie zapanować nad głodem, potrzebuje on ogromnej pomocy, kontroli z zewnątrz, ze strony otaczających go osób [8]. Podstawowymi zasadami w postępowaniu dietetycznym są ograniczenie zawartości kalorycznej posiłków do ok. 50–60% zapotrzebowania odpowiednio dla wieku, unikanie potraw o dużej zawartości tłuszczu i wysokim indeksie glikemicznym, specjalistyczne szkolenie i stałe wsparcie doświadczonego dietetyka [7]. W poradniku dla rodzin pacjentów z PWS stworzono Strategie pomocne w stosowaniu diety [8]:
1. Jedzenie tylko wyznaczonych posiłków, nic poza tym.
2. Nikt nie dokarmia dziecka, o diecie uprzedzeni są wszyscy mający kontakt z dzieckiem.
3. Zabezpieczenie dostępu do żywności w domu – „kuchnia pod kluczem”.
4. Organizując czas wolny zadbać, by nie był okazją do podjadania.
5. Zaplanowanie strategii postępowania w sytuacjach niecodziennych – uczestnictwa w przyjęciach, świętach, wycieczkach, gdzie ograniczenie diety może wymknąć się spod kontroli.
6. Niedawanie dzieciom kieszonkowego.
7. Uczenie zasad, rozróżnianie produktów dozwolonych i niedozwolonych, norm współżycia społecznego.
8. Uporządkowanie czasu – ustalone pory posiłków i ćwiczeń fizycznych.
9. Nieużywanie jedzenia jako „karty przetargowej”, nagrody.
10. Niejadanie potraw „zakazanych” przy dziecku.
Po przeanalizowaniu stylu życia rodziny opisywanej przez nas pacjentki zauważamy, że wszystkie te metody mają zastosowanie praktyczne i przestrzeganie ich stanowi klucz do sukcesu w walce z chorobą. Równie ważne jak dieta jest utrzymywanie stałego programu aktywności ruchowej, także wpisanej jako stały punkt w plan każdego dnia. Wytężone stosowanie diety i ćwiczeń fizycznych nie dawałoby jednak tak dobrych efektów w utrzymaniu prawidłowych parametrów wagowo-wzrostowych, gdyby nie współistniejąca terapia hormonem wzrostu, która okazała się przełomem w leczeniu pacjentów z PWS [8]. Liczne pozycje z piśmiennictwa wskazują na pozytywny, wielokierunkowy efekt działania hormonu wzrostu u pacjentów z PWS: zwiększenie tempa wzrastania, uzyskanie wzrostu końcowego >10c, poprawę składu ciała (stosunku beztłuszczowej do tłuszczowej masy ciała – zwiększenie ilości tkanki mięśniowej i jej siły), zwiększenie aktywności ruchowej i tolerancji wysiłku, przyspieszenie tempa przemiany materii – wzrost zapotrzebowania na energię z 50 do 75% normy dla wieku i wzrostu [5–8,10–12]. Działanie metaboliczne i wzrostowe hormonu wzrostu odbywa się w większości za pośrednictwem IGF-1, częściowo także przez bezpośredni wpływ na komórki i tkanki [6].
W skuteczności terapii zespołu PWS ważne jest jak najwcześniejsze rozpoznanie choroby, aby od pierwszych miesięcy życia rozpocząć rehabilitację poprawiającą rozwój ruchowy dziecka, od 2 roku życia stosować niskokaloryczną odpowiednio dobraną dla dziecka dietę oraz jak najwcześniej rozpocząć terapię hormonem wzrostu wzmacniającą efekt dwóch poprzednich składowych terapii zintegrowanej. Optymalny wiek rozpoczęcia terapii GH to 2–4 r.ż. [6] W piśmiennictwie znajdujemy prace dowodzące korzyści terapeutycznych wczesnej (nawet w 1 r.ż.) kwalifikacji do programu terapii GH [10,13,14], jednak ze względu na niewielką ilość danych dotyczących leczenia GH najmłodszych dzieci w Polsce zaleca się rozpoczynanie terapii GH po ukończeniu 2 r.ż. [5,8].
Terapia hormonem wzrostu może prowadzić do wystąpienia działań niepożądanych, szczególnie przy zastosowaniu zbyt dużych dawek i związanym z tym nadmiernym podwyższeniem stężenia IGF 1. W związku z tym pacjenci w trakcie leczenia wymagają monitorowania poziomu IGF 1 a także szeregu specjalistycznych badań kontrolnych [6]. Pod wpływem GH może wystąpić obniżona tolerancja glukozy [6], co przejściowo wystąpiło u opisywanej przez nas pacjentki, mogło być to jednak także związane ze zmniejszeniem kontroli nad przestrzeganiem diety przez dziecko. Często podkreślanym niebezpiecznym efektem terapii GH, szczególnie w początkowym jej stadium oraz u pacjentów z już rozwiniętą otyłością, jest ryzyko nagłego zgonu związanego z występowaniem bezdechów sennych, które mogą być pochodzenia ośrodkowego lub obturacyjnego ( skutek otyłości, osłabienia mięśni oddechowych, przerostu migdałków) [6–8, 14,15]. Opisywano epizody obturacyjnych bezdechów sennych u pacjentów, którzy przed terapią GH nie wykazywali takich zaburzeń, a hormon wzrostu prawdopodobnie spowodował u nich hipertrofię migdałków [16], a także wzrost liczby epizodów bezdechów u pacjentów prezentujących takie zaburzenia już przed włączeniem leczenia GH [17]. Opisana przez nas pacjentka zarówno przed włączeniem leczenia GH, jak i w trakcie była regularnie badana przez laryngologa, łącznie z badaniem fiberoskopowym jamy nosowo-gardłowej. Nie stwierdzono u niej przerostu migdałków, rodzice nie obserwowali zaburzeń oddychania w czasie snu.
Hormon wzrostu ma działanie anaboliczne nie tylko na mięśnie szkieletowe, ale także na mięsień sercowy, dlatego wskazane jest regularne badanie kardiologiczne pacjentów leczonych GH [6,18].
Podsumowanie/wnioski
1. Skuteczne leczenie pacjentów z PWS jest trudne i wymaga współpracy specjalistów z różnych dziedzin: endokrynologa, dietetyka, rehabilitanta, psychologa, logopedy, laryngologa, kardiologa, ortopedy.
2. Ważne jest wczesne rozpoznanie zespołu PWS, co warunkuje wczesne włączenie rehabilitacji, diety oraz leczenia GH.
3. Skuteczność zapobiegania metabolicznym powikłaniom zespołu PWS zależy od równoczesnego stosowania terapii hormonem wzrostu, rehabilitacji i diety – tzw. terapii zintegrowanej.
4. Prawdziwy sukces w leczeniu pacjentów z zespołem PWS w ogromnej mierze zależy od zaangażowania, determinacji, poświęcenia i konsekwencji ze strony ich rodziców, opiekunów.
5. Należy pamiętać o możliwych działaniach niepożądanych terapii hormonem wzrostu, poddawać pacjentów regularnym specjalistycznym badaniom kontrolnym, rozważyć korzyści i ryzyko wynikające z terapii.
Objaśnienia skrótów: PWS – Prader Willi syndrome, zespół Pradera-Willego, GH – growth hormone, hormon wzrostu, PDA – patent ductus arteriosus, przetrwały przewód tętniczy, EKG –elektrokardiogram, IM – mitral insufficiency, niedomykalność zastawki dwudzielnej,
IT – tricuspid insufficiency, niedomykalność zastawki trójdzielnej, RV – right ventricle, prawa komora, OGTT – doustny test obciążenia glukozą
Piśmiennictwo
1. Ledbetter D.H., Riccardi V.M., Airhart S.D. et al.; Deletions of chromosome 15 as a cause of the Prader-Willi syndrome; New England Journal of Medicine 1981:304 (6), 325-329
2. Nicholls R.D. et al.; Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome; Nature 1989:342, 281-285
3. Holm V.A., Cassidy S.B., Butler M.G. et al.; Prader-Willi syndrome: consensus diagnostic criteria; Pediatrics 1993:91(2), 398-402
4. Gunay-Aygun M., Schwartz S., Heeger S. et al.; The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria; Pediatrics 2001,108(5), 92
5. Spodar K., Krajewska-Walasek M.; Zespół Pradera i Willego – diagnostyka i terapia; Stand. Med. Pediatr. 2006:3, http://www.standardy.pl/pediatria/2006/5/zespol-pradera
6. ; Załącznik nr 23 do zarządzenia Nr 59/2011/DGL Prezesa NFZ z dnia 10 października 2011 roku. Terapeutyczne Programy Zdrowotne 2012. Leczenie Dzieci z zespołem Prader-Willi;
7. Mann N.P., Butler G.E.; Objawy kliniczne i postępowanie w przebiegu zespołu Pradera i Willego; Med. Prakt. Pediatr. 2011:1(73), 53-60
8. Libura M.; Moje dziecko ma zespół Pradera-Willego – jak mogę mu pomóc?; Wydawnictwo Polskie Stowarzyszenie Pomocy Osobom z Zespołem Pradera-Willego Warszawa 2007
9. Bizzari C., Rigamonti A.B., Luce A. et al.; Children with Prader-Willi syndrome exhibit more evident meal – induced responses In plasma ghrelin and peptide YY levels than obese and lean children; Eur. J. Endocrinol. 2010:162, 499-505
10. Carrel A.I., Myers S.E., Whitman B.Y. et al.; Long-Term Growth Hormone Therapy Changes the Natural History of Body Composition and Motor Function In Children with Prader-Willi Syndrome; J. Clin. Endocrinol. Metab. 2010:95(3), 1131-1136
11. Colmenares A., Pinto G., Taupin P. et al.; Efects on growth and metabolizm of growth hormone treatment for 3 years In 36 children with Prader-Willi syndrome; Horm. Res. Paediatr. 2011:75(2), 123-130
12. Sipila I., Sintonen H., Hietanen H. et al.; Long-term effects of growth hormone therapy on patients with Prader-Willi syndrome; Acta Paediatr. 2010:99 (11), 1712-1718
13. Nyunt O., Harris M, Hughes I. et al.; Benefit of early commencement of growth hormone therapy in children with Prader-Willi syndrome; J. Pediatr. Endocrinol. Metab. 2009:22(12), 1151-1158
14. Kalina-Faska B., Małecka-Tendera E.: Komentarz do: Mann N.P., Butler G.E.; Objawy kliniczne i postępowanie w przebiegu zespołu Pradera i Willego; Med. Prakt. Pediatr. 2011:1(73), 53-60
15. McCandless S. et al.; Health Supervision for Children with Prader-Willi syndrome; Pediatrics 2011:127, 195
16. Nixon G.M., Rodda Ch.P., Davey M.J. et al.; Longitudinal Association between Growth Hormone Therapy and Obstructive Sleep Apnea In a Child with Prader-Willi Syndrome; J. Clin. Endocrinol. Metab. 2011:96(1), 29-33
17. Miller J.L., Shuster J., Theriaque D. et al.; Sleep Disordered Breathing in Infants with Prader-Willi Syndrome During the First 6 Weeks of Growth Hormone Therapy: A Pilot Study; J. Clin. Sleep. Med. 2009:5(5), 448-453
18. Hauffa B.P., Knaup K., Lehmann N. et al.; Effects of growth hormone therapy on cardiac dimensions In children and adolescents with Prader-Willi syndrome; Horm. Res. Paediatr. 2011:75 (1), 56-62