Endokrynol. Ped. 12/2013;1(42):67-76
DOI: 10.18544/EP-01.12.01.1442
Zespół dysgenetycznych jąder: patogeneza i konsekwencje kliniczne
1Zakład Endokrynologii Płodności, Katedra Andrologii i Endokrynologii Płodności, Uniwersytet Medyczny w Łodzi
2Klinika Endokrynologii i Diabetologii; Instytut „Pomnik – Centrum Zdrowia Dziecka” w Warszawie
Słowa kluczowe: jądro, dysgenezja gonad, zaburzenia różnicowania płciowego, wnętrostwo, niepłodność, nowotwór jądra z komórek płciowych, ksenoestrogeny
Streszczenie
Niepełna organogeneza (dysgenezja) gonad dotyczy zwykle jąder i łączy się z zaburzeniem ich czynności hormonalnej już w życiu płodowym. Brak lub zmniejszenie wydzielania testosteronu przez płodowe komórki Leydiga prowadzi do zaburzeń różnicowania męskich wewnętrznych i zewnętrznych narządów płciowych, a także zaburzeń determinacji płci psychicznej. Z kolei nieprawidłowe wydzielanie hormonu antymüllerowskiego (AMH) przez komórki Sertoliego łączy się z przetrwaniem przewodów Müllera i obecnością żeńskich wewnętrznych narządów płciowych u osobników z męską płcią genetyczną. Dalszą konsekwencją kliniczną zaburzeń organogenezy jąder jest rozwój hipogonadyzmu hipergonadotropowego i brak płodności. Oprócz klasycznej dysgenezji gonad, ujawniającej się odwróceniem cech płciowych, istnieją prawdopodobnie jej niepełne, utajone formy. Można zaliczyć tu wnętrostwo i obniżoną płodność. Wszystkim formom dysgenezji jąder towarzyszy zwiększone ryzyko zmian nowotworowych wywodzących się z komórek płciowych. Ponieważ zaburzenia te mogą być spowodowane niepełną organogenezą jąder, zaproponowano, aby utworzyć z nich jeden zespół chorobowy nazywany zespołem dysgenetycznych jąder (ang. testicular dysgenesis syndrome, TDS). W ostatnich 50 latach obserwuje się postępujące zwiększenie częstości występowania zaburzeń należących do zespołu TDS. Uważa się, że ich przyczyną jest zanieczyszczenie środowiska naturalnego substancjami o działaniu estrogenopodobnym
Wstęp
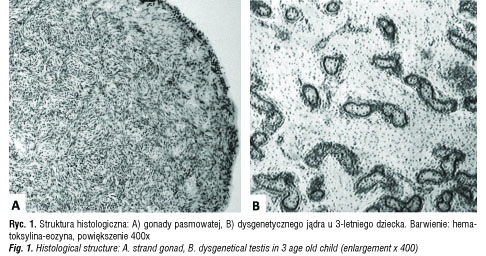
Klasycznie terminem „dysgenezja gonad” określa się różnego stopnia zahamowanie organogenezy gonad, najczęściej jąder. Histopatologicznie wyróżnia się trzy typy dysgenezji jąder: 1) czystą lub całkowitą, gdzie zamiast struktur jądra obustronnie stwierdza się pasma łącznotkankowe przypominające zrąb jajnika, 2) mieszaną, gdzie po jednej stronie znajduje się słabo rozwinięta struktura jądra,
a po drugiej pasmo łącznotkankowe i 3) częściową, kiedy stwierdza się obustronnie strukturę jądra, jednak z zaburzeniami rozwoju kanalików plemnikotwórczych [1, 2] (ryc. 1). Niepełna organogeneza jąder łączy się z zaburzeniami ich czynności hormonalnej już w życiu płodowym, czego konsekwencją są zaburzenia różnicowania i rozwoju męskich narządów płciowych oraz zaburzenia determinacji męskiej identyfikacji płciowej [3].
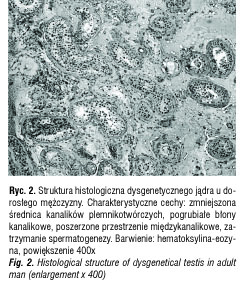
Jednak oprócz klasycznej dysgenezji jąder, ujawniającej się m.in. pełnym odwróceniem męskich cech płciowych w kierunku żeńskim (u osób z męską płcią genetyczną), istnieją jej niepełne, utajone formy, przy których nie występują zaburzenia różnicowania płciowego [4]. Skakkebaek i wsp. [5] stworzyli hipotezę, według której zaburzenia rozwojowe męskiego układu płciowego, m.in. wnętrostwo, spodziectwo, a także niepłodność spowodowana uszkodzoną spermatogenezą i nowotwory jąder wywodzące się z komórek płciowych (ang. germ cell tumours, GCT), można zaliczyć do jednej grupy, nazwanej zespołem dysgenetycznych jąder (ang. testicular dysgenesis syndrome, TDS). TDS obejmuje nieodwracalne zaburzenia, które wynikają z nieprawidłowego rozwoju jąder w okresie prenatalnym. Charakterystyczną cechą wszystkich zaburzeń zaliczanych do tego zespołu jest zwiększone ryzyko wystąpienia GCT. Różne odmiany TDS łączą podobne zmiany histopatologiczne w strukturze jąder. Kanaliki plemnikotwórcze mają zmniejszoną średnicę i zawierają niedojrzałe komórki Sertoliego, czasem same komórki Sertoliego (ang. Sertoli cell only syndrome, SCOS), ciała hialinowe i zwapnienia oraz komórki wewnątrzkanalikowego raka jądra (carcinoma in situ, CIS) [6, 7] (ryc. 2). Przestrzenie międzykanalikowe są poszerzone, a komórki Leydiga występują w nadmiernej liczbie.
Ciężkie postacie TDS (klasyczna forma dysgenezji jąder, której towarzyszą zaburzenia różnicowania narządów płciowych) występują stosunkowo rzadko: 0,77/10 tys. żywych urodzeń/rok w Europie w latach 1980–2003. W Niemczech wady narządów płciowych występują z częstością 2/10 tys. urodzeń/rok. Natomiast łagodne formy TDS są bardziej powszechne, np. wnętrostwo – 120–150/10 tys. noworodków płci męskiej/rok, spodziectwo – 7,96/10 tys. noworodków płci męskiej/rok [przegląd piśmiennictwa 8, 9]. Tendencje w obniżaniu się liczebności plemników w nasieniu odnotowano w kilku krajach europejskich oraz w USA [10, 11].
Patogeneza TDS
Obecny stan wiedzy nie pozwala na pełne zrozumienie mechanizmów patogenezy TDS. Badania kliniczne i doświadczalne potwierdziły, że za-
burzenie organogenezy jąder spowodowane różnymi przyczynami prowadzi do upośledzenia czynności komórek Sertoliego i Leydiga w okresie płodowym, co z kolei jest przyczyną zaburzeń rozwojowych męskiego układu płciowego (ZRP, ang. disorders of sex development, DSD) [12–16]. Stwierdzono, że procesy różnicowania płodowych komórek Leydiga są regulowane przez komórki Sertoliego [12, 17, 18]. Zaburzenie interakcji między tymi komórkami może być przyczyną zmniejszonej sekrecji testosteronu przez płodowe komórki Leydiga, co z kolei może prowadzić do zahamowania proliferacji komórek Sertoliego [19–21]. Liczba komórek Sertoliego zwiększa się w tym samym czasie, kiedy poziomy testosteronu są wysokie, w okresie płodowym i kilka miesięcy po urodzeniu. W jądrach dojrzałych mężczyzn liczba komórek Sertoliego jest głównym determinantem odpowiedniej wydajności wytwarzania plemników. Tak więc obniżenie poziomu testosteronu w dysgenetycznych jądrach płodowych i noworodkowych prowadzi do zmniejszenia liczby komórek Sertoliego, a w konsekwencji do zmniejszenia liczby plemników [12]. Ponadto z powodu nieprawidłowego różnicowania komórek somatycznych jądra (Sertoliego) nie wytwarzają one czynników parakrynnych i hormonów, co w konsekwencji prowadzi do braku stymulacji różnicowania płodowych komórek płciowych (gonocytów) i często do ich przemiany nowotworowej [22]. Ostatnie dane wskazują, że androgeny stymulują także ekspresję czynnika Insl3 (ang. insulin-like factor 3, INSL3), który ma istotne znaczenie dla procesu zstępowania jąder [23].
Podłożem TDS mogą być czynniki genetyczne, np. liczbowe i strukturalne aberracje chromosomów płciowych [24, 25]. Przypuszcza się także, że TDS może być związany z występowaniem specyficznej haplogrupy chromosomu Y – hp26, która jest najczęściej stwierdzana w populacji duńskiej i być może jest szczególnie wrażliwa na czynniki środowiskowe. Może to być przyczyną wysokiej zachorowalności na GCT w Danii [26].
W ostatnich 50 latach obserwuje się wzrastającą częstość występowania zaburzeń dotyczących męskiego układu płciowego. Przypuszcza się, że przyczyną tych zaburzeń mogą być zanieczyszczenia środowiska naturalnego przez substancje pochodzenia przemysłowego, zwłaszcza w regionach o wysokim poziomie rozwoju gospodarczego. Wzrasta liczba dowodów, że może to być problem globalny, chociaż stwierdza się znaczne różnice geograficzne w częstości występowania tych zaburzeń. Pojawiają się doniesienia opisujące zaburzenia czynności układu płciowego nie tylko u ludzi, ale także w świecie zwierzęcym, włączając ssaki, ptaki, ślimaki, ryby i gady. Nawet niedźwiedzie polarne na Dalekiej Północy mają problemy z rozrodem [27].
Wiele badań wykazało związek między częstością występowania zaburzeń układu rozrodczego a wzmożoną ekspozycją na czynniki środowiskowe o działaniu biologicznym naśladującym estrogeny (tzw. ksenoestrogeny), które mają także działanie antyandrogenne [8, 14, 22, 28, 29]. Ksenoestrogeny zalicza się do szerokiej grupy nazywanej w piśmiennictwie angielskim endocrine disrupting compounds (EDC) lub endocrine disruptors (ED). W nomenklaturze polskiej brakuje dobrego określenia dla EDC, a stosowane czasem sformułowanie „modulatory hormonalne” nie do końca oddaje charakter tych związków, które raczej zaburzają niż modulują gospodarkę hormonalną. Lepszym określeniem wydaje się termin „pseudohormony”.
Pseudohormony, w tym ksenoestrogeny, nie mają jednolitej struktury chemicznej. Do tej grupy zalicza się zarówno związki alifatyczne, jak
i aromatyczne, niektóre z nich zawierają w strukturze metale ciężkie lub halogeny. Przynależność poszczególnych związków do pseudohormonów determinowana jest nie przez strukturę chemiczną, ale przez sposób działania na organizmy żywe. Mogą one zaburzać biosyntezę estrogenów i androgenów, zwiększać lub zmniejszać metabolizm hormonów i zmieniać hormonalną homeostazę. Wydaje się, że ich wpływ na męski układ płciowy jest szczególnie niebezpieczny w okresie płodowym. Męski płód wytwarza białka alfa-fetoproteinę (AFP) i globulinę wiążącą steroidy płciowe (ang. sex hormone binding globulin, SHBG), które wiążą estrogeny pochodzące od matki. W ten sposób płód jest chroniony przed działaniem matczynych estrogenów. Ksenoestrogeny nie są wiązane przez te białka. Prowadzi to do ekspozycji płodów męskich na estrogenny wpływ czynników środowiska zewnętrznego [30]. Podejrzewa się, że dzięki temu mogą one uczestniczyć w patogenezie TDS oraz patogenezie GCT [22]. Podając ksenoestrogen ftalan dibutylu (dibutyl phthalate, DBP) stworzono model zwierzęcy naśladujący zmiany, jakie są obserwowane u mężczyzn z TDS [19–21]. U 60% noworodków szczura płci męskiej poddanych w okresie płodowym działaniu DBP obserwowano zaburzenia organogenezy jąder, wnętrostwo, spodziectwo i niepłodność. Opisywano zahamowanie wydzielania testosteronu, hiperplazję komórek Leydiga, obecność tych komórek wewnątrz kanalików plemnikotwórczych, występowanie wielojądrzastych gonocytów i kanalików z niedojrzałymi komórkami Sertoliego oraz opóźnienie różnicowania komórek okołokanalikowych. Innymi czynnikami, które mogą zaburzać rozwój jąder w okresie płodowym, są wewnątrzmaciczne zahamowanie wzrostu płodu, wcześniactwo, palenie papierosów przez matkę w okresie ciąży [31, 32].
Przypuszcza się, że patogeneza TDS i GCT jest wspólna. Skakkebaek [33] przedstawił hipotezę, według której wszystkie GCT, oprócz nasieniaka spermatocytarnego, wywodzą się z pierwotnych płodowych komórek płciowych, gonocytów. Uległy one przemianie nowotworowej w okresie płodowym, przetrwały aż do okresu dojrzałości płciowej w obrębie kanalików plemnikotwórczych (carcinoma in situ jądra, CIS), a następnie dały początek jawnym, inwazyjnym formom GCT. Podstawą tej teorii było stwierdzenie, że wszystkie GCT zawierają komórki o cechach morfologicznych i antygenowych podobnych do gonocytów. W ostatnich latach metodami immunohistochemicznymi zidentyfikowano wiele antygenów na powierzchni komórek CIS, takich jak: PLAP (ang. placental like alkaline phosphatase, fosfataza zasadowa typu łożyskowego) [34, 35], M2A, 43-9F, TRA-1-60 [36, 37], receptor c-kit [38], SCF (ang. stem cell factor, czynnik komórek pnia) [39], Gb3 [40], NSE (ang. neuron-specific enolase, enolasa specyficzna dla neuronów) [41], OCT3/4, NANOG, AP2γ [42–44] i inne. Większość z tych antygenów jest obecna w płodowych komórkach płciowych oraz w komórkach inwazyjnych form GCT [45, 46].
Wykazano także, że w dysgenetycznych jądrach dzieci komórki płciowe mogą być zmienione nowotworowo i wykazywać aneuploidalną zawartość DNA, typową dla GCT już we wczesnym okresie przeddojrzewaniowym [47, 48]. Znalezienie nowotworowych komórek płciowych u dzieci z zaburzeniami rozwoju płciowego potwierdza przeddojrzewaniowe pochodzenie GCT. Ponieważ stwierdzono, że w komórkach CIS oraz w płodowych i niemowlęcych komórkach płciowych występuje ekspresja tych samych białek, zaproponowano hipotezę, że inicjacja transformacji nowotworowej komórek płciowych odbywa się wcześnie w okresie płodowym. W wyniku wielu badań ustalono, że ekspresja białek płodowych jest hamowana lub opóźniona w prawidłowych gonocytach, kiedy różnicują się one do prespermatogonii [49]. Jedna z teorii mówi, że przyczyną powstania GCT mogą być mutacje genów odpowiedzialnych za postęp spermatogenezy, zlokalizowanych w regionie AZFb na ramieniu długim chromosomu Y. Produkt białkowy genów tego regionu, tzw. białko RBM (ang. RNA binding motiff, element wiążący RNA), jest obecny w prawidłowych komórkach spermatogenezy przez całe życie, począwszy od drugiego trymestru ciąży.
Natomiast nie stwierdzono ekspresji tego białka w komórkach GCT i CIS u dojrzałych płciowo mężczyzn i w jądrach dzieci z zaburzeniami organogenezy [50].
Dlaczego zjawisko to występuje tylko w niektórych przypadkach, nie jest do końca wyjaśnione. Przypuszcza się, że w dysgenetycznych jądrach przyczyną zmian nowotworowych nie są zaburzenia samych gonocytów, ale raczej ma tutaj udział zaburzenie czynności komórek somatycznych znajdujących się w otoczeniu komórek płciowych (Leydiga i Sertoliego lub ich prekursorów) [14, 51–53]. Nieprawidłowa czynność komórek somatycznych jest przyczyną obumierania większości komórek płciowych, ale niektóre z nich mają zdolność przeżycia i proliferacji. Płodowe gonocyty, które przeżyły w obrębie już wytworzonych kanalików jądra, mają lepsze warunki do przetrwania w niezmienionej, ale opóźnionej rozwojowo formie przez wiele lat jako komórki CIS. Mogą one ulec zanikowi, ale także mogą dać początek GCT w okresie dojrzewania lub późniejszym.
Istnieją hipotezy, że podwyższające się stężenie gonadotropin w okresie dojrzewania i dojrzałości płciowej u pacjentów z TDS działa na komórki CIS, wywołując ich proliferację i jest być może ostatnim ogniwem promocji nowotworu jądra.
U dzieci z dysgenezją jąder już w okresie przeddojrzewaniowym stwierdzono znamienną dodatnią korelację między liczebnością komórek CIS a stężeniem gonadotropin we krwi [54]. Postulowany jest także udział androgenów w patogenezie GCT [28]. Zwiększone działanie androgenów w okresie rozwojowym zmniejsza ryzyko nowotworowe. U kobiet rasy czarnej obserwuje się większe stężenia
androgenów w okresie ciąży w porównaniu z kobietami rasy białej. Wiązane z tym zjawiskiem jest rzadkie występowanie GCT u Afrykanów, ale za to częste występowanie raka prostaty [55].
Konsekwencje kliniczne
Prawidłowa czynność hormonalna płodowych jąder warunkuje organogenezę wewnętrznych i zewnętrznych męskich narządów płciowych. Różnicowanie narządów płciowych w kierunku męskim odbywa się pomiędzy 6 a 20 tygodniem życia płodowego pod wpływem wytwarzanych przez jądra androgenów, a także hormonu antymüllerowskiego (AMH). Testosteron stymuluje przekształcanie przewodów śródnerczowych (Wolffa) w wewnętrzne narządy płciowe męskie, tj. najądrza, nasieniowody, pęcherzyki nasienne i brzuszną część gruczołu krokowego. Do powstania zewnętrznych narządów płciowych męskich z wzgórka płciowego i zatoki moczowo-płciowej niezbędny jest dihydrotestosteron (DHT), 3-krotnie silniejszy androgen powstający z testosteronu przy udziale enzymu 5-alfa reduktazy w tkankach obwodowych. Z kolei AMH wywołuje zanik zawiązków żeńskich wewnętrznych narządów płciowych (przewody przyśródnerczowe Müllera) u płodów męskich [56].
Prawdopodobnie testosteron wydzielany przez jądra w okresie okołoporodowym ma znaczenie w determinacji rozwoju struktur mózgu odpowiedzialnych za męską identyfikację płciową. Wykazano, że raz wykształcone poczucie przynależności płciowej jest nieodwracalne, a obustronna kastracja w okresie przeddojrzewaniowym nie zmienia wcześniej zdeterminowanego kierunku rozwoju identyfikacji płciowej [57].
Brak lub zmniejszenie wydzielania testosteronu przez komórki Leydiga prowadzi do zaburzeń rozwoju płciowego (ZRP). Nieprawidłowe wydzielanie AMH przez komórki Sertoliego łączy się z przetrwaniem przewodów Müllera i obecnością żeńskich wewnętrznych narządów płciowych u osobników z męską płcią genetyczną [58].
W przypadku całkowitego braku organogenezy gonad obustronnie obecne są pasma tkanki łącznej włóknistej, przypominające zrąb jajnika, ale bez pęcherzyków jajnikowych. Narządy płciowe wewnętrzne i zewnętrzne, a także identyfikacja płciowa są tu zwykle typu żeńskiego, gdyż gonada taka nie wydziela testosteronu.
W przypadku słabego różnicowania gonad w kierunku jąder stwierdza się strukturę gonady męskiej, ale z zaburzeniami rozwoju kanalików plemnikotwórczych. Zaburzenie czynności hormonalnej takich jąder może być różnie nasilone (brak do prawie prawidłowej czynności) i w związku z tym narządy płciowe mogą być żeńskie lub nie w pełni zmaskulinizowane. Płeć psychiczna w takich przypadkach jest trudna do przewidzenia w okresie noworodkowym [59].
Klinicznie najważniejszymi objawami ciężkich postaci dysgenezji jąder są: 1) lokalizacja gonad w jamie brzusznej lub kanałach pachwinowych, 2) obojnacze lub żeńskie zewnętrzne narządy płciowe u osobnika z chromosomem Y lub jego częścią w kariotypie, 3) słabo rozwinięte męskie i żeńskie lub tylko żeńskie narządy płciowe wewnętrzne, 4) niskie stężenie testosteronu we krwi i obniżona w stosunku do rówieśników rezerwa wydzielnicza testosteronu w teście z hCG przed i w okresie dojrzewania, 5) podwyższone stężenia LH i FSH już w okresie przeddojrzewaniowym i bardzo wysokie u dorosłych (tab. I).

Wady rozwojowe męskiego układu płciowego występują coraz częściej. W Polsce stanowią one ok. 13% wad wrodzonych i zajmują trzecie miejsce po wadach układu krążenia i układu kostnego [60]. Najczęstszą wadą jest spodziectwo (lokalizacja ujścia cewki moczowej przesunięta w kierunku krocza na brzusznej stronie trzonu prącia). Wiele danych wskazuje także na coraz częstsze występowanie wnętrostwa [61].
Tymczasem fenotyp TDS może się ujawniać u zdrowych pod innymi względami mężczyzn w trakcie lub po dojrzewaniu płciowym, dlatego te przypadki mogą być niezauważone w okresie dziecięcym (tab. II). Prawdopodobnie najczęstszym objawem TDS jest uszkodzenie spermatogenezy (azoospermia lub dużego stopnia oligozoospermia <5 mln/ml plemników w nasieniu) i związana z tym niepłodność. Badania Carlsen i wsp. [10] wykazały pogarszające się od ok. 50 lat parametry jakości nasienia. Autorzy przeprowadzili tzw. metaanalizę na podstawie publikacji z lat 1938–1990 dotyczących płodności mężczyzn. Stwierdzili, że liczba plemników w tym okresie stopniowo zmniejszała się ze średnio 113 mln/ml w latach 40. do 66 mln/ml w latach 80. XX wieku. W późniejszych badaniach opisywano także pogarszanie się morfologii (cech budowy) plemników, a także ich ruchliwości. Takiej tendencji nie zaobserwowano jednak w Finlandii, gdzie średnia liczebność plemników w nasieniu wynosi 124 mln/ml i jest największa w Europie.

W Danii 20% młodych zdrowych mężczyzn ma koncentrację plemników poniżej 20 mln/ml (dolna granica normy zalecanej przez WHO w 1999 r., według nowszych zaleceń z 2010 r. – 15 mln/ml) i ok. 40% ma mniej plemników niż 40 mln/ml, co uznaje się za granicę, poniżej której występuje liniowa zależność między liczebnością plemników i częstością występowania ciąż [62]. Analiza wyników badania nasienia przeprowadzona w Poradni Andrologii i Endokrynologii Płodności przy Uniwersyteckim Szpitalu Klinicznym nr 3 w Łodzi nie wykazała pogarszania się liczebności plemników u polskich mężczyzn w latach 1982–2000. Stwierdzono jednak zmniejszanie się odsetka plemników z prawidłową morfologią.
Gorszy stan nabłonka plemnikotwórczego wiąże się z innymi parametrami dysgenezji jąder, m.in. zmniejszoną średnicą kanalików plemnikotwórczych, pogrubiałą błoną kanalikową, poszerzonymi przestrzeniami międzykanalikowymi, obecnością kanalików z niedojrzałymi komórkami Sertoliego [63]. W badaniu USG stwierdza się niehomogenną strukturę jąder i liczne mikrozwapnienia. Objętość jąder z TDS jest zmniejszona (poniżej 12 ml).
Należy jednak podkreślić, że nie wszystkie przypadki gorszych parametrów nasienia, a przez to obniżonego potencjału płodności, oraz wnętrostwa i zaburzeń rozwoju męskich narządów płciowych są konsekwencją zaburzeń rozwoju jąder i TDS [64]. Przyczyna męskiej niepłodności może mieć podłoże czysto genetyczne, np. mutacje odcinka AZF (ang. azoospermic factor) na ramieniu długim chromosomu Y. Nieprawidłowe wyniki badania nasienia mogą być także spowodowane niedrożnością przewodów najądrza lub nasieniowodów, stanem zapalnym w układzie płciowym lub trybem życia. Z kolei zaburzenia zstępowania jąder czy wady rozwojowe układu płciowego mogą być spowodowane brakiem wrażliwości na androgeny lub niedoborem 5α-reduktazy. TDS może jednak odgrywać istotną rolę w gwałtownym zwiększaniu się problemów związanych z męskim układem płciowym, czego nie można tłumaczyć jedynie zmianami genetycznymi, a raczej wpływem środowiska [65].
We wszystkich zaburzeniach składających się na TDS występuje zwiększone ryzyko rozwoju GCT [13, 14]. Pacjenci z „klasyczną” dysgenezją jąder mają największe ryzyko rozwoju GCT [47, 54, 66]. Słowikowska-Hilczer i wsp. [67] stwierdzili, że najczęściej, bo w 91% przypadków, zmiany nowotworowe występują przy częściowej dysgenezji gonad (najsłabiej wyrażonym zaburzeniu organogenezy jąder), rzadziej w mieszanej (77% przypadków), a najrzadziej w czystej (23%). Częstość występowania jawnych GCT zwiększa się w tempie 2–3,5%/rok w krajach skandynawskich i ok. 5%/rok w Polsce i Niemczech. Większe tempo wzrostu częstości zachorowań na te nowotwory występuje u mężczyzn młodych, poniżej 30 roku życia. Najczęściej nowotwory jąder stwierdzane są w Danii (ok. 12 nowych przypadków/100 tys. mężczyzn/rok). Nowotwory jąder są obecnie najczęstszymi nowotworami u młodych mężczyzn rasy białej [26, 68].
Podsumowanie
Objawy kliniczne TDS pojawiają się w różnych formach i z różnym nasileniem, dlatego są diagnozowane i leczone przez lekarzy różnych specjalności, m.in. pediatrów, endokrynologów, urologów, ginekologów, seksuologów, onkologów. Stwierdzono, że występowanie kilku objawów TDS u jednej osoby może być częstsze niż dotychczas przypuszczano na podstawie oceny pojedynczych objawów. Wskazuje to na potrzebę umieszczania problematyki TDS w programach szkoleń lekarzy różnych specjalności, aby uwzględniali oni te zaburzenia w diagnostyce i zdawali sobie sprawę z ich konsekwencji, przede wszystkim z zagrożenia nowotworowego.
Praca finansowana z funduszy projektu badawczego MNiSW nr N N407 277339
Piśmiennictwo
1. Nezelof C.; Gonadal dysgenesis and agenesis: anatomical expression; Bull. Assos. Anat. 1991:75, 43-45
2. Berkovitz G.D., Seeherunvong T.; Abnormalities of gonadal differentiation; Baillieres Clin. Endocrinol. Metab. 1998:12, 133-142
3. Słowikowska-Hilczer J., Kula K.; Kliniczne konsekwencje zaburzeń organogenezy jądra i obwodowego działania steroidów płciowych; End. Diab. Chor. Przem. Mat. 2000:6, Supl. 1, 51-56
4. Wohlfart-Veje C., Main K.M., Skakkebaek N.E.; Testicular dysgenesis syndrome: foetal origin of adult reproductive problems; Clin. Endocrinol. 2009:71, 459-465
5. Skakkebaek N.E., Rajpert-DeMeyts E., Main K.M.; Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects; Hum. Reprod. 2001:16, 972-978
6. Hoei-Hansen C.E., Holm M., Rajpert-De Meyts E. et al.; Histological evidence of testicular dysgenesis in contralateral biopsies of 218 patients with testicular germ cell cancer; J. Pathol. 2003:200, 370-374
7. Rajpert-DeMeyts E., Hoei-Hansen C.E.; From gonocytes to testicular cancer: the role of impaired gonadal development; Ann. N. York Acad. Sci. 2007:1120, 168-180
8. Słowikowska-Hilczer J.; Xenobiotics with estrogen or antiandrogen action – disruptors of the male reproductive system; Centr. Europ. J. Med. 2006:3, 205-227
9. Thyen U., Lanz K., Holterhus P.M., Hiort O.; Epidemiology and initial management of ambiguous genitalia at birth in Germany; Horm. Res. 2006:66, 195-203
10. Carlsen E., Giwercman A., Keiding N. et al.; Evidence for decreasing quality of semen during past 50 years; Brit. Med. J. 1992:305, 467-471
11. Swan S.H., Elkin E.P., Fenster L.; The question of declining sperm density revisited: an analysis of 101 studies published 1934-1996; Environ. Health Perspect. 2000:108, 961-966
12. Sharpe R.M., McKinnell C., Kivlin C. et al.; Proliferation and functional maturation of Sertoli cells, and their relevance to disorders of testis function in adulthood; Reproduction 2003:125, 769-784
13. Skakkebaek N.E., Holm M., Hoei-Hansen C. et al.; Association between testicular dysgenesis syndrome (TDS) and testicular neoplasia: Evidence from 20 adult patients with signs of maldevelopment of the testis; APMIS 2003:111, 1-11
14. Skakkebaek N.E.; Testicular dysgenesis syndrome: new epidemiological evidence; Int. J. Androl. 2004:27, 189-191
15. Andersson A.-M., Jørgensen N., Frydelund-Larsen L. et al.; Impaired Leydig cell function in infertile men: A study of 357 idiopathic infertile men and 318 proven fertile controls; J. Clin. Endocrinol. Metab. 2004:89, 3161-3167
16. Joensen U.N., Jorgensen N., Rajpert-DeMeyts E. et al.; Testicular Dysgenesis Syndrome and Leydig cell function; Basic Clin. Pharmacol. Toxicol. 2008:102, 155-161
17. Risbridger G.P., Kerr J.B., De Kretser D.M.; Evaluation of Leydig cell function and gonadotrophin binding in unilateral and bilateral cryptorchidism: evidence for local control of Leydig cell function by the seminiferous tubule; Biol. Reprod. 1981:24, 534-540
18. Booth J.D., Merriam G.R., Clark R.V. et al.; Evidence for Leydig cell dysfunction in infertile men with a selective increase in plasma follicle-stimulating hormone; J. Clin. Endocrinol. Metab. 1987:64, 1194-1198
19. Mylchreest E., Cattley R.C., Foster P.M.D.; Male reproductive tract malformations in rats following gestational and lactational exposure to di(n-butyl) phthalate: an antiandrogenic mechanisim?; Toxicol. Sci. 1998:43, 47-60
20. Mylchreest E., Sar M., Wallace D. et al.; Fetal testosterone insufficiency and abnormal proliferation of Leydig cells and gonocytes in rats exposed to di(n-butyl) phthalate; Reprod. Toxicol. 2002:16, 19-28
21. Fisher J.S., Macpherson S., Marchetti N. et al.; Human “testicular dysgenesis syndrome”: a possible model using in-utero exposure of the rat to dibutyl phthalate; Hum. Reprod. 2003:18, 1383-1394
22. Rajpert-DeMeyts E.; Developmental model for the pathogenesis of testicular carcinoma in situ: genetic and environmental aspects; Hum. Reprod. Update 2006:12, 303-323
23. McKinnell C., Sharpe R.M., Mahood K. et al.; Expression of insulin-like factor 3 protein in the rat testis during fetal and postnatal development and in relation to cryptorchidism induced by in utero exposure to di (n-butyl) phthalate; Endocrinol. 2005:146, 4117-4126
24. Lim H.N., Freestone S.H., Romero D. et al.; Candidate genes in complete and partial XY sex reversal: Mutation analysis of SRY, SRY-related genes and FTZ-F1; Mol. Cell. Endocrinol. 1998:140, 51-58
25. Müller J., Ritzen E.M., Ivarsson S.A. et al.; Management of males with 45,X/46,XY gonadal dysgenesis; Horm. Res. 1999:52, 11-1
26. Møller H., Evans H.; Epidemiology of gonadal germ cell cancer in males and females; APMIS 2003:111, 43-46
27. Vos J.G., Dybing E., Greim H.A. et al.; Health effects on endocrine-disrupting chemicals on wildlife, with special reference to the European situation; Crit. Rev. Toxicol. 2000:30, 71-133
28. Rajpert-DeMeyts E., Skakkebaek N.E.; The possible role of sex hormones in the development of testicular cancer; Eur. Urol. 1993:23, 54-59
29. Sharpe R.M.; Hormones and testis development and the possible adverse effects of environmental chemicals; Toxicol. Lett. 2001:120, 221-232
30. Bonde J.P., Giwercman A.; Occupational hazards to male fecundity; Reprod. Med. Rev. 1995:4, 59-73
31. Jensen T.K., Jorgensen N., Punab M. et al.; Association of in utero exposure to maternal smoking with reduced semen quality and testis size in adulthood: a cross-sectional study of 1770 young men from the general population in five European countries; Am. J. Epidemiol. 2004:159, 49-58
32. Main K.M., Jensen R.B., Asklund C. et al.; Low birth weight and male reproductive function; Horm. Res. 2006:65, 116-122
33. Skakkebaek N.E.; Possible carcinoma-in-situ of the testis; Lancet 1972:9, 516-517
34. Jacobsen G.K., Nørgaard-Pedersen B.; Placental alkaline phosphatase in testicular germ cell tumours and in carcinoma-in-situ of the testis; APMIS 1984:92, 323-329
35. Rajpert-DeMeyts E., Bartkova J., Samson M. et al.; The emerging phenotype of the testicular carcinoma in situ germ cell; APMIS 2003:111, 267-279
36. Giwercman A., Andrews P.W., Jørgensen N. et al.; Immunohistochemical expression of embryonal marker TRA-1-60 in carcinoma in situ and germ cell tumours of the testis; Cancer 1993:72, 1308-1314
37. Giwercman A., Cantell L., Marks A.; Placental-like alkaline phosphatase as a marker of carcinoma-in-situ of the testis. Comparison with monoclonal antibodies M2A and 43-9F; APMIS 1991:99, 586-594
38. Rajpert-DeMeyts E., Skakkebaek N.E.; Expression of the c-kit protein product in carcinoma in situ and invasive germ cell tumours; Int. J. Androl. 1994:17, 85-92
39. Strohmeyer T., Reese D., Press M. et al.; Expression of the c-kit proto-oncogene and its ligand stem cell factor (SCF) in normal and malignant human testicular tissue; J. Urol. 1995:153, 511-515
40. Kang J.L., Rajpert-DeMeyts E., Wiels J. et al.; Expression of the glycolipid globotriaosylceramide (Gb3) in testicular carcinoma in situ; Virchows Arch. 1995:426, 369-374
41. Kang J.L., Rajpert-DeMeyts E., Skakkebaek N.E.; Immunoreactive neuron-specific enolase (NSE) is expressed in testicular carcinoma-in-situ; J. Pathol. 1996:178, 161-165
42. Rajpert-DeMeyts E., Hanstein R., Jorgensen N. et al.; Developmental expression of POU5FI (OCT-3/4) in normal and dysgenetic human gonads; Hum. Reprod. 2004:19, 1338-1344
43. Hoei-Hansen C.E., Nielsen J.E., Almstrup K. et al.; Transcription factor AP-2gamma is a developmentally regulated marker of testicular carcinoma in situ and germ cell tumors; Clin. Cancer Res. 2004:10, 8521-8530
44. Hoei-Hansen C.E., Almstrup K., Nielsen J.E. et al.; Stem cell pluripotency factor NANOG is expressed in human fetal gonocytes, testicular carcinoma in situ and germ cell tumors; Histopathol. 2005:47, 48-56
45. Rajpert-DeMeyts E., Kvist M., Skakkebaek N.E.; Heterogeneity of expression of immunohistochemical tumour markers in testicular carcinoma in situ: pathogenetic relevance; Virchows Arch. 1996:428, 133-139
46. Andrews P.W.; Teratocarcinomas and human embryology: pluripotent human EC cell lines. Review article; APMIS 1998:106, 158-167
47. Słowikowska-Hilczer J., Szarras-Czapnik M., Kula K.; Testicular pathology in 46,XY dysgenetic male pseudohermaphroditism: an approach to pathogenesis of testis cancer; J. Androl. 2001:22, 781-792
48. Chemes H., Fuzulin P.M., Venara M.C. et al.; Early manifestation of testicular dysgenesis in children: pathological phenotypes, karyotype correlations and precursor stages of tumour development; APMIS 2003:111, 12-23
49. Rajpert-DeMeyts E., Jorgensen N., Muller J. et al.; Prolonged expression of the c-kit receptor in germ cells of intersex fetal testes.; J. Pathol. 1996:178, 166-169
50. Schreiber L., Lifschitz-Mercer B., Paz G. et al.; Double immunolabeling by the RBM and the PLAP markers for identifying intratubular (in situ) germ cell neoplasia of the testis; Int. J. Surg. Pathol. 2003:11, 17-20
51. Cools M., Stoop H., Kersemaekers A.M.F, et al.; Gonadoblastoma arising in undifferentiated gonadal tissue within dysgenetic gonads; J. Clin. Endocrinol. Metab. 2006:91, 2404-2413
52. Looijenga L., Gillis A.J.M., Stoop H. et al.; Dissecting the molecular pathways of (testicular) germ cell tumour pathogenesis; from initiation to treatment-resistance; Int. J. Androl. 2011:34, e234-e251
53. Looijenga L., Hersmus R., Leeuw H. et al.; Gonadal tumours and DSD; Best Pract. Res. Clin. Endocrinol. Metab. 2010:24, 291-310
54. Słowikowska-Hilczer J.; Nuclear DNA content and proliferative potential of human carcinoma in situ cells in testes of intersex children; Folia Histochem. Cytobiol. 2001:39, 167-168
55. English P.B., Goldberg D.E., Wolff C. et al.; Parental and birth characteristics in relation to testicular cancer risk among males born between 1960 and 1995 in California (United States); Cancer Causes Control 2003:14, 815-825
56. Conte F.A., Grumbach M.M.; Zaburzenia determinacji i różnicowania płci. W: Endokrynologia ogólna i kliniczna (Greenspan F.S. i Gardner D.G., wyd. pol. Lewiński A.); Czelej Lublin 2004, 545-584
57. Swaab D.F., Hofman M.A.; Sexual differentiation of the human brain: A historical perspective; Prog. Brain Res. 1990:61, 361-374
58. Rey R.A., Belville C., Nihoul-Fekete C. et al.; Evaluation of gonadal function in 107 intersex patients by means of serum anti-Müllerian hormone measurement; J. Clin. Endocrinol. Metab. 1999:84, 627-631
59. Kula K., Słowikowska-Hilczer J.; Konsekwencje zaburzeń działania hormonów płciowych w obrębie centralnego układu nerwowego: zmiany behawioralne, anatomiczne i czynnościowe; Neurol. Neurochir. Pol. 2003:4, 19-39
60. Latos-Bieleńska A., Materna-Kiryluk A., Krawczyński M.R. et al.; Genital malformations in Poland – data from the Polish Registry of Congenital Malformations; Horm. Res. 1999:5, 78
61. Thonneau P.F., Candia P., Mieusset R.; Cryptorchidism: Incidence, risk factors, and potential role of environment; an update; J. Androl. 2003:24, 155-162
62. Bonde J.P., Ernst E., Jensen T.K. et al.; Relation between semen quality and fertility: a population-based study of 430 first-pregnancy planners; Lancet 1998:352, 1172-1127
63. Guminska A., Oszukowska E., Kuzanski W. et al.; Less advanced testicular organogenesis is associated with a higher incidence of germ cell neoplasia; Int. J. Androl. 2010:33, 153-162
64. Jorgensen N., Rajpert-DeMeyts E., Main K.M., Skakkebaek N.; Testicular dysgenesis syndrome comprises some but not all cases of hypospadias and impaired spermatogenesis; Int. J. Androl. 2010:33, 298-303
65. Toppari J., Virtanen H.E., Main K.M., Skakkebaek N.E.; Cryptorchidism and hypospadias as a sign of testicular dysgenesis syndrome (TDS): Environmental connection; Birth Defects Res. Clin. Mol. Teratol. 2010:88, 910-919
66. Dieckmann K.P., Pichlmeier U.; Clinical epidemiology of testicular germ cell tumours; World J. Urol. 2004:22, 2-14
67. Słowikowska-Hilczer J., Romer T.E., Kula K.; Neoplastic potential of germ cells in relation to disturbances of gonadal organogenesis and changes in karyotype; J. Androl. 2003:24, 270-278
68. Shah M.N., Devesa S.S., Zhu K. et al.; Trends in testicular germ cell tumours by ethnic group in the United States; Int. J. Androl. 2007:30, 206-213